
Abstract
Begomoviruses (Geminiviridae) are economically important, high consequence plant viruses transmitted by members of the Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) cryptic species complex. One of the most economically significant begomoviruses is tomato yellow leaf curl virus (TYLCV), which is efficiently transmitted by B. tabaci Middle East–Asia Minor 1 (MEAM1), one of the most destructive agricultural pests globally. The purpose of this study is to determine the gene expression profiles of non-viruliferous and viruliferous MEAM1 after TYLCV acquisition access feeding. Adult whiteflies were fed on healthy or TYLCV infected tomato plants for 12, 36, or 60 hours, followed by a 12-hour gut clearing period on collard, a TYLCV non-host. Differential expression analyses (read mapping and transcript quantification) from RNA-seq data derived from ribodepleted total RNA were performed to determine gene expression patterns associated with TYLCV acquisition. In total, 37 differentially expressed genes were identified, including two horizontally acquired from plants. Elucidating the transcriptional response of MEAM1 to virus acquisition can inform the development of novel genomics-assisted whitefly transmitted virus management strategies.
Similar content being viewed by others

Background & Summary
Since its description as a pest of tobacco in Greece1, the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae) has been a persistent scourge to field and greenhouse-grown agricultural crops, fibre commodities, and ornamental plants around the world2. In the United States, the 1980’s invasion of B. tabaci B biotype3, now often referred to as the Middle East–Asia Minor 1 (MEAM1)4 (also mitotype B or Bemisia argentifolii Bellows & Perring5,6), resulted in hundreds of millions of dollars in economic losses. Plant damage associated with rapid increases in the whitefly population, extreme polyphagy7, physiological disorders due to direct feeding8,9, and, most seriously, virus transmission10, were attributed to the new invader. Approximately 40 years later, B. tabaci continues to pose substantial problems for agriculturalists and subsistence farmers throughout the world, mainly due to the transmission of viruses, including begomoviruses (family Geminiviridae) which severely limit yield in numerous vegetable11, root12, and fibre crops13.
Begomoviruses are dicotyledonous plant viruses with single-stranded, mono- or bipartite circular genomes approximately 2.7 kilobases in length14. Viruses in this genus are spread exclusively by members of the B. tabaci cryptic species complex in a persistent, circulative manner15. Differential transmission of certain begomoviruses has been reported for different species within the complex16,17,18. The route viral particles take within the whitefly vector is complex19. The whitefly uses its stylets to ingest virions from phloem sap of an infected host, which then pass through the food canal, cibarium, and esophagus before entering the midgut filter chamber. The filter chamber is the main site where virions concentrate and are translocated into the haemolymph20,21,22. Once in the haemolymph, viral particles are protected en route to the primary salivary glands by complexing with a GroEL chaperone protein manufactured by the whitefly endosymbiotic bacterium Hamiltonella in MEAM123,24 and Arsenophonus in Asia II-1 (also referred to as biotype/mitotypes K, P, ZHJ2, PCG-1, SY, PK14,25). Virions are then internalized into the salivary glands and finally egested back into the plant phloem, where the cycle starts anew.
One of the most serious viral pathogens transmitted by B. tabaci is tomato yellow leaf curl virus (TYLCV)26, a monopartite begomovirus that causes tomato yellow leaf curl disease (TYLCD)27. The onset and severity of TYLCD symptoms are highly variable and dependent on the tomato cultivar, timing of inoculation relative to plant senescence, and environmental conditions. Typical TYLCD symptoms include upward curling and yellowing of leaflet margins, reduced size of leaflets, stunting, aborted blooms, reduced fruit size, and irregular fruit ripening8,28. Complete yield losses due to TYLCD are not uncommon27. Due to this notoriety, TYLCV is one of the most studied begomoviruses transmitted by B. tabaci, and developing effective management strategies to reduce TYLCV-induced economic losses is an active area of research29,30,31.
A promising research avenue to reduce the deleterious effects of TYLCV on tomato production is the adoption of genomics-based technologies. This has manifested as the identification of genetic loci that confer resistance to TYLCV and their integration into stable tomato breeding lines32,33,34, as well as the development of RNA interference (RNAi) technologies targeting genes in the vector that affect different life history parameters (e.g., longevity, fecundity, etc.) and virus transmission35,36,37,38. Target genes for RNAi may be identified through transcriptomic studies that evaluate the response of the whitefly to begomovirus acquisition. This is accomplished by measuring the number of differentially expressed genes (DEGs) between viruliferous and non-viruliferous whiteflies after they are allowed to feed on a host with or without the pathogen of interest. Coupled with a well-annotated genome assembly39,40,41, the DEGs can be identified, and their functions determined, to select the best candidates for downstream control applications (e.g., RNAi, CRISPR)42,43.
Several studies have examined differential gene expression in B. tabaci following begomovirus acquisition44,45,46,47,48,49,50,51,52,53,54,55,56,57, but relatively few have investigated the transcriptional response of B. tabaci MEAM1 to TYLCV acquisition. Those that have reported substantially different numbers of DEGs, which is most likely a result of key methodological differences including the use of propagative material (i.e., infected plant cuttings) instead of potted plants49, the sequencing of only one tissue type (e.g., gut tissue)48, and the implementation of a gut-clearing step following the TYLCV acquisition access period (AAP)54. In this study, we examined the transcriptional response of B. tabaci to TYLCV after three AAPs (12, 36, and 60 h) on potted tomato followed by a gut-clearing step on a TYLCV non-host, collard (Brassica oleracea). The gut-clearing step was implemented to minimize any potential indirect effects that feeding on infected plant tissue has on the vector44,54, thereby increasing the likelihood that changes in gene expression profiles between viruliferous and non-viruliferous whiteflies directly reflect virus-vector interactions. We also took a novel approach to preparing the sequenced RNA-seq libraries as compared to previous studies of the vector transcriptional response to begomovirus acquisition. To date, rRNA depleted RNA-seq libraries have not been reported in similar studies of whitefly-virus interactions. Previous studies have demonstrated that gene quantification variation is dependent on RNA sample preparation, with rRNA depletion able to capture more unique and rare transcripts than poly(A) selection58,59,60. Such transcripts include those encoding replication-dependent histone proteins61 as well as long-noncoding RNAs62. This dataset will be useful to researchers interested in studying novel B. tabaci genetic targets for developing enhanced genomics-informed whitefly and whitefly-transmitted virus management.
Methods
Whitefly colony
A colony of B. tabaci MEAM1 was established in 2022 from adult whiteflies collected on field grown zucchini (Cucurbita pepo L.) in the research fields of the Coastal Research and Education Center in Charleston, South Carolina, USA. Whiteflies were then reared on collard (Brassica oleracea var. acephala) in a greenhouse (26 ± 4 °C) at the USDA-ARS, U. S. Vegetable Laboratory, with fresh collard plants added to the colony as needed.
Plant inoculations
To produce viruliferous whiteflies for use in plant inoculations, approximately 20 mating pairs of the MEAM1 colony maintained on collard were placed in clip cages and transferred to visibly symptomatic tomato plants (Solanum lycopersicum L., cultivar ‘Moneymaker’) infected with TYLCV for a 72-hour acquisition access period (AAP). Healthy tomato plants with at least three fully expanded true leaves were then subjected to feeding by approximately 20 mating pairs of non-viruliferous (healthy) or TYLCV-viruliferous (virus) whiteflies in clip cages for a 72-hour inoculation access period (IAP) to generate mock-inoculated (whitefly-exposed) and TYLCV-infected plants for use in experiments 3–4 weeks later. Plant infection status was assessed with end-point PCR before the initiation of feeding assays using primer pair KL14-324 (5′-CTTCGACAGCCCATACAGCA-3′) and KL14-325 (5′-GAGGGCCCACCAATAACTGT-3′) designed by Dr. Daniel Hasegawa in the laboratory of Dr. Kai-Shu Ling (USDA, ARS, U.S. Vegetable Laboratory, Charleston, SC, USA).
Feeding assays
To acquire tomato-acclimated, age-specific (staged) whiteflies for use in experiments, older collard leaves from the whitefly colony bearing 4th instar whitefly nymphs, with adult whiteflies removed, were excised and added to cages containing virus-free tomato plants for a five-day emergence period. On day five, the emerged adult whiteflies were aspirated into standard insect collection vials and released onto the mock-inoculated or TYLCV-infected tomato plants. Approximately 1,500 adult whiteflies (≤ five days post-emergence) were added to each experimental plant, which were held in separate BugDorm cages (60 × 60 × 60 cm; MegaView Science Co., Ltd., Taichung, Taiwan) in an insectary maintained at 26 ± 4 °C, 45 ± 15% RH, and a natural light/dark cycle (14:10 L:D). Whiteflies were allowed to feed for three acquisition access periods (AAPs): 12, 36, and 60 hours. At the end of each AAP, approximately 500 adult whiteflies from each experimental cage were transferred to collard, a non-host of TYLCV, for a 12-hr gut clearing period. The gut clearing step was added to ensure that differential expression was due to virus ingestion and acquisition rather than a response to feeding on TYLCV-infected plant tissue. Following gut clearing, all whiteflies were collected and frozen at −80 °C until RNA extraction. The experiment was replicated three times, and each replication consisted of one plant per treatment.
RNA extraction, Illumina library preparation, and RNA sequencing
Total RNA was extracted from whiteflies using TRIzol® with the PureLink® RNA Mini Kit following the manufacturer’s recommended protocol, except that initial sample lysis was conducted in 100 µL TRIzol® using an ice-cold pestle and the optional on-column DNase treatment protocol was performed. Illumina library preparation and RNA sequencing were performed at the Genomics Core Facility of Michigan State University (East Lansing, MI, USA). Whitefly and bacterial (symbiont) ribosomal RNA (rRNA) was depleted from samples using the QIAseq FastSelect rRNA Fly and 5S/16S/23S kits, respectively (Qiagen, Hilden, Germany). Stranded RNA-seq libraries were prepared using the Illumina Stranded Total RNA Library Preparation Ligation Kit (Illumina, Inc., San Diego, CA, USA) with Integrated DNA Technologies Unique Dual Index adapters following the manufacturer’s recommendations, except that half volume reactions were used. Library quality control and quantification was conducted using a combination of Qubit dsDNA HS (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and Agilent 4200 TapeStation HS DNA1000 assays (Agilent Technologies, Santa Clara, CA, USA). The libraries were pooled in equimolar proportions and quantified using an Invitrogen Collibri Quantification qPCR kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Sequencing was performed on an Illumina NovaSeq6000 sequencing system. The pool was loaded onto one lane of an Illumina S4 flow cell, and sequencing was performed in a 2×150bp paired-end format using a NovaSeq v1.5, 300 cycle reagent kit. Base calling was done by Illumina Real Time Analysis (RTA) v3.4.4, and the output of RTA was demultiplexed and converted to FASTQ format with Illumina Bcl2fastq (v2.20.0).
RNA-seq read processing, mapping, and differential expression analyses
Raw reads from the 18 RNA-seq libraries (six treatments with three replicates) were processed with fastp63 (v.0.12.4) to remove sequencing adapters, trim low-quality bases, and remove reads less than 75 base pairs (bp) in length. The cleaned reads were then aligned to the MEAM1 mitochondrial genome (NCBI Reference Sequence: NC_006279.1), three bacterial endosymbiont genomes (Candidatus Portiera aleyrodidarum, Candidatus Hamiltontella defensa, and Rickettsia sp. available at http://www.whiteflygenomics.org/ftp/MEAM1/endosymbionts/), and Release 138.1 of the SILVA rRNA database (SSU and LSU NR 99 Ref sequences; accessed on 26 June 2023) using HISAT264 (v.2.2.1). The aligned reads were filtered out, and the remaining reads were aligned to the MEAM1 nuclear genome assembly39 with STAR65 (v.2.7.10b) using the splice site information included in the MEAM1 general feature format file at http://www.whiteflygenomics.org/ftp/MEAM1/v1.2/MEAM1_v1.2.gff3.gz. The resulting SAM files were converted to BAM format with samtools66 (v.1.17). Differential expression (DE) analyses on read- and gene-level count matrices were performed with DESeq. 267 (v.1.40.2) in R68. The count matrix of sequencing reads unambiguously mapped to a gene (hereafter referred to as read mapping; RM) was generated using the BAM files mentioned previously. The gene-level count matrix (hereafter referred to as transcript quantification; TQ) was created with Salmon69 (v.1.6.0) and imported to R using the tximport package70 (v.1.28.0). In both RM and TQ analyses, genes with an adjusted p-value < 0.1 and minimum fold change of 1.2 were considered as differentially expressed. The illustrations in Fig. 1 were created using the package ggplot271 (v.4.4.1) in R.

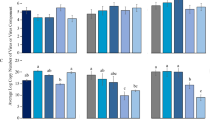
Bar chart of the number of differentially expressed genes (DEGs) at each acquisition access period (AAP) identified by read mapping (RM) and transcript quantification (TQ).
On average, 32.5 million cleaned RNA-seq reads were generated per library (N = 18), and 96.1% of the cleaned reads in each library aligned to the MEAM1 nuclear genome assembly (Table 1). Approximately four million more RNA-seq reads were generated per library in viruliferous (36.6 million) vs non-viruliferous whiteflies (32.6 million).
Thirty-seven DEGs were identified across all time points and types of analysis (Fig. 1; Table 2). Thirteen and 26 DEGs were identified by RM and TQ, respectively. Two genes (Bta05741, Bta05749) were identified as differentially expressed in both analyses, and no genes were differentially expressed at more than one time point. In RM analyses, 10 genes were upregulated, and 3 genes were downregulated. Two of the genes upregulated at 72 hours (Bta13103, Bta13961) were horizontally acquired from plants72. In TQ analyses, 8 genes were upregulated, and 18 genes were downregulated.
Data Records
The data associated with this project have been accessioned with the National Center for Biotechnology Information (NCBI) under BioProject number PRJNA1096732: Transcriptomics of viruliferous and non-viruliferous Bemisia tabaci MEAM173. Illumina RNA-seq reads have been deposited in the NCBI Sequence Read Archive under accession numbers SRR28578498–SRR28578515, and the BioSamples used to generate the RNA-seq reads have been assigned BioSample accession numbers SAMN40761531– SAMN40761536.
Technical Validation
Total RNA quantification was performed using a DeNovix DS-11 FX spectrophotometer/fluorometer (DeNovix Inc., Wilmington, DE, USA) using 1 µL of total RNA. RNA integrity was assessed on a 2100 Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA, USA) at the Molecular Analytics Core, part of the Department of Regenerative Medicine and Cell Biology at the Medical University of South Carolina (Charleston, SC, USA). Cohorts of six (non-viruliferous) and 10 (viruliferous) whiteflies from each replicate and time point were collected and individually extracted to screen for the absence or presence of TYLCV. All whiteflies in the non-viruliferous treatments tested negative for TYLCV at each time point. In the viruliferous treatments, 90%, 93%, and 97% of the whiteflies at 24, 48, and 72 hours, respectively, tested positive for TYLCV.
Select up- and downregulated genes from the RM and TQ DEG analyses were validated using a QIAcuity One digital PCR (dPCR) system (QIAGEN, Hilden, Germany) (Supplementary Table 1). Full-length coding sequences of each gene in the B. tabaci MEAM1 genome (version 1.2; Chen et al. 2016), downloaded at http://www.whiteflygenomics.org/ftp/MEAM1/v1.2/MEAM1_CDS_v1.2.fa.gz, were used to design primers with Primer3web74 (version 4.1.0) at default parameters (https://primer3.ut.ee/). Primers for each DEG are listed in Supplementary Table 2. dPCR Reactions consisted of 5 µL EvaGreen master mix (QIAGEN, Hilden, Germany), 0.6 µL 10 µM forward and reverse primers, 1.5 µL cDNA at 500 ng/µL, and 7.3 µL nanopure water. Reactions were performed in 24 or 96 well nanoplates with 8.5k partitions per well. Thermocycling consisted of an initial 2 min denaturation at 95 °C, 40 cycles at 95 °C for 30 s and 60 °C for 60 s, and a final extension for 10 min at 35 °C. Imaging was performed at an exposure duration of 200 ms and gain of 6, and the threshold for positive partitions was set at 50 RFU. The expression of each gene was normalized to the whitefly gene α-tubulin, a housekeeping gene.
Data availability
Datasets are available through the NCBI repository BioProject number PRJNA1096732 at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1096732.
Code availability
A text file with example commands used to reproduce the analyses is included as a supplement.
References
Gennadius, P. Disease of tobacco plantations in the Trikonia. The aleurodid of tobacco. [In Greek]. Ellenike Georgia 5, 1–3 (1889).
Stansly, P. A. & Naranjo, S. E. Bemisia: Bionomics And Management Of A Global Pest (Springer 2010).
Costa, H. S. & Brown, J. K. Variation in biological characteristics and esterase patterns among populations of Bemisia tabaci, and the association of one population with silverleaf symptom induction. Entomol. Exp. Appl. 61, 211–219, https://doi.org/10.1111/j.1570-7458.1991.tb01553.x (1991).
De Barro, P. J., Liu, S.-S., Boykin, L. M. & Dinsdale, A. B. Bemisia tabaci: A Statement of Species Status. Annu. Rev. Entomol. 56, 1–19, https://doi.org/10.1146/annurev-ento-112408-085504 (2011).
Bellows, T. S., Perring, T. M., Gill, R. J. & Headrick, D. H. Description of a Species of Bemisia (Homoptera: Aleyrodidae). Ann. Entomol. Soc. Am. 87, 195–206, https://doi.org/10.1093/aesa/87.2.195 (1994).
Brown, J. K. in Bemisia: Bionomics And Management Of A Global Pest (eds. Stansly, P. A. & Naranjo, S. E.) Chapter 2: Phylogenetic biology of the Bemisia tabaci sibling species group https://doi.org/10.1007/978-90-481-2460-2_2 (Springer, 2010).
Mound, L. A. & Halsey, S. H. Whitefly of the world: a systematic catalogue of the Aleyrodidae (Homoptera) with host plant and natural enemy data, https://doi.org/10.5962/bhl.title.118687 (British Museum and Wiley, 1978).
Schuster, D. J., Mueller, T. F., Kring, J. B. & Price, J. F. Relationship of the sweetpotato whitefly to a new tomato fruit disorder in Florida. HortScience 25, 1618–1620, https://doi.org/10.21273/HORTSCI.25.12.1618 (1990).
Yokomi, R. K., Hoelmer, K. A. & Osborne, L. S. Relationships between the sweetpotato whitefly and the squash silverleaf disorder. Phytopathology 80, 895–900, https://doi.org/10.1094/Phyto-80-895 (1990).
Markham, P. G., Bedford, I. D., Liu, S. & Pinner, M. S. The transmission of geminiviruses by Bemisia tabaci. Pestic. Sci. 42, 123–128, https://doi.org/10.1002/ps.2780420209 (1994).
Adkins, S., Webb, S. E., Achor, D., Roberts, P. D. & Baker, C. A. Identification and characterization of a novel whitefly-transmitted member of the family Potyviridae isolated from cucurbits in Florida. Phytopathology 97, 145–154, https://doi.org/10.1094/PHYTO-97-2-0145 (1997).
Tembo, M., Mataa, M., Legg, J., Chikoti, P. C. & Ntawuruhunga, P. Cassava mosaic disease: incidence and yield performance of cassava cultivars in Zambia. J. Plant Pathol. 99, 681–689, https://www.jstor.org/stable/44687139 (2017).
Naranjo, S. E., Chu, C.-C. & Henneberry, T. J. Economic injury levels for Bemisia tabaci (Homoptera: Aleyrodidae) in cotton: impact of crop price, control costs, and efficacy of control. Crop Prot. 15, 779–788, https://doi.org/10.1016/S0261-2194(96)00061-0 (1997).
Fiallo-Olivé, E. et al. ICTV virus taxonomy profile: Geminiviridae 2021. J. Gen. Virol. 102, 001696, https://doi.org/10.1099/jgv.0.001696 (2021).
Mehta, P., Wyman, J. A., Nakhla, M. K. & Maxwell, D. P. Transmission of Tomato yellow leaf curl geminivirus by Bemisia tabaci (Homoptera: Aleyrodidae). J. Econ. Entomol. 87, 1291–1297, https://doi.org/10.1093/jee/87.5.1291 (1994).
Chi, Y. et al. Differential transmission of Sri Lankan cassava mosaic virus by three cryptic species of the whitefly Bemisia tabaci complex. Virology 540, 141–149, https://doi.org/10.1016/j.virol.2019.11.013 (2020).
Fiallo-Olivé, E., Pan, L.-L., Liu, S.-S. & Navas-Castillo, J. Transmission of begomoviruses and other whitefly-borne viruses: dependence on the vector species. Phytopathology 110, 10–17, https://doi.org/10.1094/PHYTO-07-19-0273-FI (2020).
Gautam, S. et al. Differential transmission of Old and New World begomoviruses by Middle East-Asia Minor 1 (MEAM1) and Mediterranean (MED) cryptic species of Bemisia tabaci. Viruses 14, 1104, https://doi.org/10.3390/v14051104 (2022).
Czosnek, H., Hariton-Shalev, A., Sobol, I., Gorovits, R. & Ghanim, M. The incredible journey of begomoviruses in their whitefly vector. Viruses 9, 273, https://doi.org/10.3390/v9100273 (2017).
Hunter, W. B., Hiebert, E., Webb, S. E. & Tsai, J. H. Location of gemniviruses in the whitefly, Bemisia tabaci (Homoptera: Aleyrodidae). Plant Dis. 82, 1147–1151, https://doi.org/10.1094/PDIS.1998.82.10.1147 (1998).
Brown, J. K. & Czosnek, H. Whitefly transmission of plant viruses. Adv. Bot. Res. 36, 65–100, https://doi.org/10.1016/S0065-2296(02)36059-2 (2002).
Czosnek, H., Ghanim, M. & Ghanim, M. The circulative pathway of begomoviruses in the whitefly Bemisia tabaci —insights from studies with Tomato yellow leaf curl virus. Ann. Appl. Biol. 140, 215–231, https://doi.org/10.1111/j.1744-7348.2002.tb00175.x (2002).
Morin, S. et al. A GroEL homologue from endosymbiotic bacteria of the whitefly Bemisia tabaci is implicated in the circulative transmission of Tomato yellow leaf curl virus. Virology 256, 75–84, https://doi.org/10.1006/viro.1999.9631 (1999).
Gottlieb, Y. et al. The transmission efficiency of Tomato yellow leaf curl virus by the whitefly Bemisia tabaci is Correlated with the presence of a specific symbiotic bacterium species. J. Virol. 84, 9310–9317, https://doi.org/10.1128/jvi.00423-10 (2010).
Rana, V. S., Singh, S. T., Priya, N. G., Kumar, J. & Rajagopal, R. Arsenophonus GroEL interacts with CLCuV and is Llocalized in midgut and salivary gland of whitefly B. tabaci. PLOS ONE 7, e42168, https://doi.org/10.1371/journal.pone.0042168 (2012).
Czosnek, H. et al. Isolation of tomato yellow leaf curl virus, a geminivirus. Phytopathology 78, 508–512, https://doi.org/10.1094/Phyto-78-508 (1988).
Moriones, E. & Navas-Castillo, J. in Bemisia: Bionomics And Management Of A Global Pest (eds. Stansly, P. A. & Naranjo, S. E.) Chapter 8. Tomato yellow leaf curl disease epidemics, https://doi.org/10.1007/978-90-481-2460-2_8 (Springer, 2010).
Polston, J. E., McGovern, R. J. & Brown, L. G. Introduction of Tomato yellow leaf curl virus in Florida and implication for the spread of this and other geminiviruses of tomato. Plant Dis. 83, 984–988. https://doi.org/10.1094/PDIS.1999.83.11.984.
Polston, J. E. & Lapidot, M. in Tomato Yellow Leaf Curl Virus Disease (ed. Czosnek, H.) Chapter 2. Management of Tomato yellow leaf curl virus: US and Israel perspectives https://doi.org/10.1007/978-1-4020-4769-5_15 (Springer, 2007).
Dhaliwal, M. S., Jindal, S. K., Sharma, A. & Prasanna, H. C. Tomato yellow leaf curl virus disease of tomato and its management through resistance breeding: a review. J. Hortic. Sci. Biotech. 95, 425–444, https://doi.org/10.1080/14620316.2019.1691060 (2020).
Li, F., Qiao, R., Yang, X., Gong, P. & Zhou, X. Occurrence, distribution, and management of tomato yellow leaf curl virus in China. Phytopathol. Res. 4, 28, https://doi.org/10.1186/s42483-022-00133-1 (2022).
Yang, Y., Sherwood, T. A., Patte, C. P., Hiebert, E. & Polston, J. E. Use of Tomato yellow leaf curl virus (TYLCV) Rep gene sequences to engineer TYLCV resistance in tomato. Phytopathology 94, 490–496, https://doi.org/10.1094/PHYTO.2004.94.5.490 (2004).
Pereira-Carvalho, R. C. et al. Recessive resistance derived from tomato cv. Tyking-Limits drastically the spread of tomato yellow leaf curl virus. Viruses 7, 2518–2533, https://doi.org/10.3390/v7052518 (2015).
Gill, U. et al. Ty-6, a major begomovirus resistance gene on chromosome 10, is effective against Tomato yellow leaf curl virus and Tomato mottle virus. Theor. Appl. Genet. 132, 1543–1554, https://doi.org/10.1007/s00122-019-03298-0 (2019).
Upadhyay, S. K. et al. RNA interference for the control of whiteflies (Bemisia tabaci) by oral route. J. Biosci. 36, 153–161, https://doi.org/10.1007/s12038-011-9009-1 (2011).
Luan, J.-B., Ghanim, M., Liu, S.-S. & Czosnek, H. Silencing the ecdysone synthesis and signaling pathway genes disrupts nymphal development in the whitefly. Insect Biochem. Mol. Biol. 43, 740–746, https://doi.org/10.1016/j.ibmb.2013.05.012 (2013).
Hariton Shalev, A., Sobol, I., Ghanim, M., Liu, S.-S. & Czosnek, H. The whitefly Bemisia tabaci Knottin-1 gene is implicated in regulating the quantity of tomato yellow leaf curl virus ingested and transmitted by the insect. Viruses 8, 205, https://doi.org/10.3390/v8070205 (2016).
Ibrahim, A. B., Monteiro, T. R., Cabral, G. B. & Aragão, F. J. L. RNAi-mediated resistance to whitefly (Bemisia tabaci) in genetically engineered lettuce (Lactuca sativa). Transgenic Res. 26, 613–624, https://doi.org/10.1007/s11248-017-0035-0 (2017).
Chen, W. et al. The draft genome of whitefly Bemisia tabaci MEAM1, a global crop pest, provides novel insights into virus transmission, host adaptation, and insecticide resistance. BMC Biol. 14, 110, https://doi.org/10.1186/s12915-016-0321-y (2016).
Xie, W. et al. Genome sequencing of the sweetpotato whitefly Bemisia tabaci MED/Q. GigaScience 6, gix018, https://doi.org/10.1093/gigascience/gix018 (2017).
Chen, W. et al. Genome of the African cassava whitefly Bemisia tabaci and distribution and genetic diversity of cassava-colonizing whiteflies in Africa. Insect Biochem. Molec. 110, 112–120, https://doi.org/10.1016/j.ibmb.2019.05.003 (2019).
Heu, C. C., McCullough, F. M., Luan, J. & Rasgon, J. L. CRISPR-Cas9-Based Genome Editing in the Silverleaf Whitefly (Bemisia tabaci). CRISPR J. 3, 89–96, https://doi.org/10.1089/crispr.2019.0067 (2020).
Shelby, E. A. et al. Debugging: strategies and considerations for efficient RNAi-mediated control of the whitefly Bemisia tabaci. Insects 11, 723, https://doi.org/10.3390/insects11110723 (2020).
Luan, J. et al. Global analysis of the transcriptional response of whitefly to Tomato yellow leaf curl China virus reveals the relationship of coevolved adaptations. J. Virol. 85, 3330–3340, https://doi.org/10.1128/jvi.02507-10 (2011).
Wang, B. et al. MicroRNA profiling of the whitefly Bemisia tabaci Middle East-Aisa Minor I following the acquisition of Tomato yellow leaf curl China virus. Virol. J. 13, 20, https://doi.org/10.1186/s12985-016-0469-7 (2016).
Kaur, N. et al. Transcriptome analysis of the whitefly, Bemisia tabaci MEAM1 during feeding on tomato infected with the crinivirus, Tomato chlorosis virus, identifies a temporal shift in gene expression and differential regulation of novel orphan genes. BMC Genomics 18, 370, https://doi.org/10.1186/s12864-017-3751-1 (2017).
Kaur, N. et al. Differences in gene expression in whitefly associated with CYSDV-infected and virus-free melon, and comparison with expression in whiteflies fed on ToCV- and TYLCV-infected tomato. BMC Genomics 20, 654, https://doi.org/10.1186/s12864-019-5999-0 (2019).
Geng, L. et al. Transcriptome profiling of whitefly guts in response to Tomato yellow leaf curl virus infection. Virol. J. 15, 14, https://doi.org/10.1186/s12985-018-0926-6 (2018).
Hasegawa, D. K. et al. Comparative transcriptome analysis reveals networks of genes activated in the whitefly, Bemisia tabaci when fed on tomato plants infected with Tomato yellow leaf curl virus. Virology 513, 52–64, https://doi.org/10.1016/j.virol.2017.10.008 (2018).
Ding, T.-B., Li, J., Chen, E.-H., Niu, J.-Z. & Chu, D. Transcriptome Profiling of the Whitefly Bemisia tabaci MED in Response to Single Infection of Tomato yellow leaf curl virus, Tomato chlorosis virus, and Their Co-infection. Front. Physiol. 10, 302, https://doi.org/10.3389/fphys.2019.00302 (2019).
Kilot, A., Kontsedalov, S., Lebedev, G., Czosnek, H. & Ghanim, M. Combined infection with Tomato yellow leaf curl virus and Rickettsia influences fecundity, attraction to infected plants and expression of immunity-related genes in the whitefly Bemisia tabaci. J. Gen. Virol. 100, 721–731, https://doi.org/10.1099/jgv.0.001233 (2019).
Li, M., Zhao, J. & Su, Y.-L. Transcriptome analysis of gene expression profiles of Tomato yellow leaf curl virus-infected whiteflies over different viral acquisition access periods. Insects 11, 297, https://doi.org/10.3390/insects11050297 (2020).
Yue, H. et al. Integrated analysis of microRNA and mRNA transcriptome reveals the molecular mechanism of Solanum lycopersicum response to Bemisia tabaci and Tomato chlorosis virus. Front. Microbiol. 12, 693574, https://doi.org/10.3389/fmicb.2021.693574 (2021).
Mugerwa, H. et al. Differential transcriptional responses in two old world Bemisia tabaci cryptic species post acquisition of old and new world begomoviruses. Cells 11, 2060, https://doi.org/10.3390/cells11132060 (2022).
Nekkanti, A. et al. Transcriptomic changes of Bemisia tabaci Asia II 1 induced by chilli leaf curl virus trigger infection and circulation in its vector. Front. Microbiol. 13, 890807, https://doi.org/10.3389/fmicb.2022.890807 (2022).
Zhao, J. et al. Changes in gene expression of whiteflies, Bemisia tabaci MED feeding on tomato plants infected by one of the criniviruses, Tomato chlorosis virus through transcriptome analysis. Int. J. Genomics 2023, 807812, https://doi.org/10.1155/2023/3807812 (2023).
Chen, T., Jia, Y., Chen, J. & Qi, G. Comparative transcriptome analysis of whiteflies raised on cotton leaf curl Multan virus-infected cotton plants. Front. Vet. Sci. 11, 1417590, https://doi.org/10.3389/fvets.2024.1417590 (2024).
Cui, P. et al. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 96, 259–265, https://doi.org/10.1016/j.ygeno.2010.07.010 (2010).
Guo, Y. et al. RNAseq by total RNA library identifies additional RNAs compared to poly(A) RNA library. BioMed Res. Int. 2015, 862130, https://doi.org/10.1155/2015/862130 (2015).
Zhao, S., Zhang, Y., Gamini, R., Zhang, B. & von Schack, D. Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: polyA+ selection versus rRNA depletion. Sci. Rep. 8, 4781, https://doi.org/10.1038/s41598-018-23226-4 (2018).
Marzluff, W. F., Wagner, E. J. & Duronio, R. J. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet. 9, 843–854, https://doi.org/10.1038/nrg2438 (2008).
Yang, L., Duff, M. O., Graveley, B. R., Carmichael, G. G. & Chen, L.-L. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 12, R16, https://doi.org/10.1186/gb-2011-12-2-r16 (2011).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. https://www.R-project.org/.
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419, https://doi.org/10.1038/nmeth.4197 (2017).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses from RNA-seq: transcript-level estimates improve gene-level inferences [version 2; peer review: 2 approved]. F1000Research 4, 1521, https://doi.org/10.12688/f1000research.7563.2 (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis https://ggplot2.tidyverse.org/ (Springer, 2016).
Gilbert, C. & Maumus, F. Multiple horizontal acquistitions of plant genes in the whitefly Bemisia tabaci. Genome Biol. Evol. 14, evac141, https://doi.org/10.1093/gbe/evac141 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP500144 (2025).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115, https://doi.org/10.1093/nar/gks596 (2012).
Acknowledgements
We thank Dr. Daniel Hasegawa (USDA-ARS) for comments on an initial draft of the manuscript that improved its quality. The first author is a participant of the Oak Ridge Institute for Science and Education (ORISE) Agricultural Research Service (ARS) Research Participation Program, supported by the USDA-ARS, U. S. Vegetable Laboratory in Charleston, SC, USA. This work has been funded by the USDA Agricultural Research Service project number 6080-22000-030-000-D. The mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA. The USDA is an equal opportunity provider and employer. This article was written without the use of artificial intelligence.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.A.A. Data curation: Z.L. Methodology: Z.L. and S.A.A. Experimentation: Z.L. Project administration and supervision: S.A.A. and A.M.S. Writing - original draft: Z.L. Writing – revised draft: Z.L., A.M.S. and S.A.A. All authors have agreed to the publication of the final version of this document.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lahey, Z., Simmons, A.M. & Andreason, S.A. Gene expression differences in Bemisia tabaci following acquisition of an Old World begomovirus. Sci Data 13, 103 (2026). https://doi.org/10.1038/s41597-025-06417-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06417-3
