
Abstract
As a protandrous hermaphrodite with natural male-to-female sex change, maroon clownfish (Premnas biaculeatus) serves as an ideal model organism for investigating sequential hermaphroditism. However, genomic resources for this interesting species remain scarce, thereby limiting in-depth research on its unique biological traits. In this study, we generated the first telomere-to-telomere (T2T) gap-free genome assembly of maroon clownfish by integrating multi-platform sequencing data, including MGI short reads, PacBio HiFi long reads, ONT ultra-long reads, and Hi-C sequencing data. The final haplotypic genome spans 884.39 Mb, with all sequences successfully anchored onto 24 chromosomes. This assembly is highly contiguous, with a contig N50 of 37.98 Mb. Comprehensive genomic characterization revealed the precise localization of telomeric repeats and centromeric region within each chromosome. Independent quality assessments, such as QV of 71.01, CRAQ score of 98.98%, and BUSCO completeness of 99.98%, confirmed good assembly accuracy. Additionally, alignment of ONT ultra-long and PacBio HiFi reads to the assembly yielded a high mapping rate exceeding 99%, further validating a good assembly integrity. Repetitive elements constituted 33.51% (296.37 Mb) of the assembled genome, and a total of 24,556 protein-coding genes were annotated. This high-quality T2T genome assembly will not only provide a valuable genetic resource to advance related research in comparative genomics, population genetics, molecular breeding, and functional genomics of maroon clownfish, but also lay a solid foundation for resolving molecular mechanisms underlying its protandrous reproductive strategy.
Similar content being viewed by others

Background & Summary
Maroon clownfish (Premnas biaculeatus), also named as spinecheek anemonefish, is a marine teleost under the order Perciformes and the family Pomacentridae. However, it is the only clownfish species that does not fall within the genus Amphiprion1. As a representative of coral reef ecosystems, maroon clownfish is distinguished by its vivid body coloration, species-specific behaviors, and its obligate symbiotic relationship with sea anemones2. These traits make it not only a globally popular ornamental fish but also a classic model organism for investigating ecological and molecular mechanisms of marine symbiosis3,4.
Evolution of sex change strategies in fish is tightly associated with their mating systems, as these strategies directly impact reproductive fitness5,6. Previous field observations and experimental studies have consistently demonstrated that, to maximize reproductive output under the constraints of social structure (e.g., dominance hierarchy), some fish species have evolved a genetic feature of sex change7. Sex change and hermaphroditic strategies in bony fishes are taxonomically and functionally diverse, and they can be categorized into four primary types based on the direction and timing of sexual differentiation: protogyny (female-first sex change), protandry (male-first sex change), bidirectional sex change (flexible transition between sexes), and synchronous hermaphroditism (simultaneous possession of functional male and female gonads)8. Representative species include Asian seabass (Lates calcarifer)9, black seabream (Acanthopagrus schlegelii)10, and orange-red pygmygoby (Trimma okinawae)11. Within the family Pomacentridae, fishes of the genera Amphiprion and Premnas are typically protandrous hermaphrodites, which initially develop as functional males and can later undergo a transition to females. In contrast, some species of the genus Dascyllus display protogynous sex change, wherein individuals first appear as females and subsequently shift to males6. Despite sharing similarities in reproductive behaviors (e.g., egg guarding) and parental care strategies with other Pomacentridae species, these genera possess highly specialized mating systems, rendering them ideal models for investigating the evolution of sex change and the structure of social behaviors.
Maroon clownfish is the only anemonefish species within the genus Premnas. It exhibits strict host specificity to the bubble-tip anemone (Entacmaea quadricolor)12. Certain studies have demonstrated that maroon clownfish holds a competitive advantage over other organisms inhabiting Entacmaea quadricolor13. Compared with other anemonefish species, maroon clownfish possesses a unique social structure. That is to say, it typically exists solely in monogamous pairs, rather than in social groups that include immature subadults. Breeding females are generally twice the size of breeding males14. Like other anemonefishes, maroon clownfish is a protandrous hermaphrodite, and its social structure is characterized by female dominance15.
In our present study, we combined MGI short-read, PacBio HiFi long-read, ONT (Oxford Nanopore Technologies) ultra-long, and Hi-C sequencing data to construct a high-fidelity telomere-to-telomere (T2T) genome assembly of maroon clownfish. This assembly was rigorously assessed for quality, and its key genomic features were characterized. This gap-free reference genome assembly will not only facilitate population genetic research and evolutionary study, but also provide an important genetic resource for molecular breeding and investigating molecular mechanisms of sex change in this economically important fish.
Methods
Sample collection
The maroon clownfish used in this study (Fig. 1A) was obtained from a local base of the South China Sea Fisheries Research Institute (SCSFRI), Chinese Academy of Fishery Sciences (CAFS), in Shenzhen city, Guangdong province, China. It had a body length of 4.6 cm and body weight of 6.5 g. For whole-genome sequencing, muscle was dissected from the individual and subjected to multi-platform sequencing, including MGI short-read, PacBio HiFi long-read, ONT ultra-long-read, and Hi-C sequencing technologies. Additionally, transcriptome sequencing (RNA-seq) was performed using four distinct tissue samples (gill, eye, muscle and skin) (Table 1). All sampling procedures and experimental workflows were conducted in compliance with the guidelines established by the Animal Ethics Committee of SCRFRI, CAFS (No. nhdf2025-30).

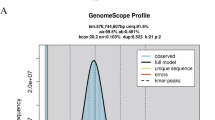
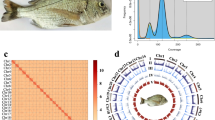
Maroon clownfish and its whole-genome sequence distribution. (A) A morphological image of the sequenced fish. (B) A k-mer (21-mer) distribution curve for estimation of the genome size.
DNA extraction and genome sequencing
Genomic DNA (gDNA) was extracted from the muscle tissue using a QIAamp DNA Mini kit (Qiagen, Valencia, CA, USA) in accordance with the manufacturer’s protocols16. Quality of the extracted gDNA was evaluated using three complementary approaches. Fragment size was determined via 0.75% agarose gel electrophoresis, molecular integrity was assessed by an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and absolute quantification was performed using a Qubit Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
For the MGI sequencing, a paired-end library with an insert size of 350 bp was constructed using a MGIEasy Universal DNA Library Preparation kit (MGI, Shenzhen, China). Sequencing was then conducted on a DNBSEQ-T7 platform (MGI) to generate 67.56 Gb of paired-end reads. These raw reads were filtered using fastp v0.12.617 with the following parameters: -n 0 -f 5 -F 5 -t 5 -T 5 -q 20. This filtering step removed adapter sequences, reads with excessive Ns, and low-quality bases (Phred quality score < 20), resulting in 59.92 Gb of clean reads (Table 1) that were used for subsequent data error correction and genome size estimation.
For the PacBio HiFi sequencing, approximately 10 μg of high-quality gDNA was used to construct a SMRTbell library following the standard protocol provided with the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, Menlo Park, CA, USA). The library was sequenced on a PacBio Sequel II System using the circular consensus sequencing (CCS) technology. HiFi reads were generated using CCS v6.0.018 with the optimized parameter “-min-passes 3”. This process yielded a total of 101.76 Gb of HiFi reads with a N50 of 17,331 bp (Table 1).
Two ultra-long read libraries were prepared following Oxford Nanopore Technologies (ONT) standard protocols and sequenced on a PromethION platform (Oxford Nanopore Technologies Co., Littlemore, Oxford, UK). Raw ONT reads were quality-filtered using NanoFilt v2.8.019 to remove those reads with a quality value (QV) < 7. After filtering, 72.04 Gb of clean ultra-long reads were retained, which had an average length of 40,256 bp and a N50 of 62,144 bp (Table 1).
For the high-throughput chromosome conformation capture (Hi-C) sequencing, a Hi-C library was constructed using a GrandOmics Hi-C kit (GrandOmics, Wuhan, Hubei, China) according to the manufacturer’s protocol. This library was sequenced on a DNBSEQ-T7 platform (MGI) using the 150-bp paired-end sequencing mode to generate 106.47 Gb of raw data. These raw reads were filtered using fastp v0.12.617 to remove adapters and low-quality sequences, resulting in 104.07 Gb of clean data (Table 1) for subsequent chromosome-level genome assembly.
RNA extraction and transcriptome sequencing
Total RNA was extracted from four tissues (Table 1) separately according to a standard Trizol protocol (Invitrogen, Frederick, MD, USA), followed by purification with a Qiagen RNeasy Mini Kit (Qiagen, Germantown, MD, USA). RNA concentration and integrity were measured using a NanoDrop 8000 Spectrophotometer (Thermo Fisher Scientific) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), respectively. Only those RNA samples with OD260/280 ≥ 1.8 and RNA integrity ≥ 7.0 were selected for transcriptome sequencing. RNA was used for construction of a cDNA library followed the manufacture’s guideline, which was then sequenced on a HiSeq X Ten platform (Illumina, San Diego, CA, USA). A total of 48.07 Gb of transcriptome raw data were generated (Table 1), which aided in annotation of protein-coding genes and prediction of gene structures.
Genome-size estimation and construction of a T2T genome assembly
To estimate the genome size of maroon clownfish, k-mer counting was performed using Jellyfish v2.2.1020 with a k-mer length of 21. The analysis parameters were configured as follows: -m 21 -s 10 G -C. A k-mer frequency histogram generated by Jellyfish was used as the input for GenomeScope v2.021, a tool designed to infer genome-wide genetic characteristics from k-mer distribution patterns. This analytical workflow allowed for sequence-based estimation of genome features prior to de novo assembly, eliminating potential biases introduced by assembly artifacts. As shown in Fig. 1B, the genome size of maroon clownfish was calculated to be approximately 842.2 Mb. Additional key genomic characteristics derived from this analysis include an estimated heterozygosity of 0.387%.
Primary contigs were initially constructed via de novo assembly of PacBio HiFi reads and ONT ultra-long reads using Hifiasm v0.19.822 with default parameters. Subsequent to contig generation, the purge_dups v1.2.523 pipeline (--low 65 --mid 115 --high 150 --min-length 1000) was applied to filter out haplotypic duplications and heterozygous redundant sequences from the initial de novo assembly. This quality control step resulted in a genome assembly with a total length of 884.09 Mb.
Using this preliminary genome assembly as a reference, Hi-C clean reads were utilized to perform chromosome-level scaffolding. The workflow was implemented as follows: (1) Hi-C clean reads were mapped to the assembled contigs using Bowtie2 v2.2.524 with the following parameter settings: --very-sensitive -L 20 --score-min L,-0.6,-0.2 --end-to-end. (2) The HiC-Pro v2.8.1 pipeline25 was applied to detect Hi-C ligation products, with only valid paired-end reads retained for downstream analysis to ensure high reliability of interaction signals. (3) Based on the valid Hi-C reads, the preliminary contig assembly was clustered into chromosome groups, ordered, and oriented to construct chromosome-level scaffolds using Juicer v1.526 and 3D-DNA v3.027. Juicebox v1.11.0828 was employed to visualize the chromosome-level assembly. Manual adjustments were further conducted to correct misclusters, misorders, or misorientations in the candidate chromosome assemblies, ensuring good consistency with Hi-C interaction patterns.
To eliminate residual gaps in the chromosome-level assembly, corrected ultra-long ONT reads were used for gap filling. Two complementary tools were implemented, including TGS-GapCloser v1.2.129 with optimized parameters: --min_match 1000 --min_nread 3, and LR_Gapcloser v1.030 with the parameter settings: -t 35 -m 1000000 -v 500. The final gap-free genome assembly of maroon clownfish has a total length of 884.39 Mb (Table 2), with all sequences anchored onto 24 chromosomes (Chr; Fig. 2). The longest Chr is 45.43 Mb, while the shortest Chr is 26.18 Mb (Table 3). The number of assembled chromosomes (haplotypic n = 24) is consistent with the previously established diploid karyotype (2n = 48) of this species31.

T2T genome assembly of maroon clownfish. (A) Genome-wide chromatin interactions at a 500-kb resolution. Color blocks represent corresponding interactions, with various strengths from white (low) to red (high). (B) A Circos plot of the main genomic features. From outside to inside the details include (1) the 24 chromosomes, (2) gene density, (3) GC content, (4) repetitive sequences density, and (5) a colinear relationship among chromosomes of the maroon clownfish genome assembly. Note that the density calculation window is set as 100 kb.
Identification of centromere and telomere sequences
Telomeres were annotated by searching for the conserved telomeric repeat motif (CCCTAA/TTAGGG) at both terminal regions of each chromosome using the Telomere-to-Telomere (T2T) Toolkit quarTeT v1.1.132. To annotate centromeres in maroon clownfish, we first identified repetitive sequences using TRF v4.0.433 and RepeatMasker v4.0.634, generating a comprehensive transposable element (TE) annotation file. This TE annotation was then used as an input for quarTeT v1.1.132 to predict candidate centromeric intervals based on the enrichment of centromere-associated repetitive elements. Collectively, our annotations confirmed that the assembled genome contains a complete set of 24 centromeres and 48 telomeres (Fig. 3), consistent with the previously established diploid karyotype for this species31.

Genome-wide localization of repetitive elements (REs), telomeres, and centromeres. Triangles at both ends of each chromosome represent the telomere regions, and the gully area within each chromosome stands for a centromere region.
Annotation of repeat elements
To annotate repetitive elements (REs) in the maroon clownfish genome, tandem repeats were identified using Tandem Repeats Finder (TRF) v4.0.433 and Genome-wide Microsatellite Analyzing Tool Package (GMATA) v2.235. Specifically, TRF was employed to detect simple sequence repeats (SSRs), while GMATA was applied to characterize all tandem repeat families across the entire assembled genome, ensuring comprehensive coverage of tandemly repeated sequences.
For transposable element (TE) annotation, a combined strategy of homology-based and de novo prediction was adopted to minimize false negatives and improve annotation accuracy. In the former approach, RepeatMasker v4.0.6 and RepeatProteinMask v4.0.634 were utilized: RepeatMasker aligned the assembled genome against the RepBase database (v26.04) to identify known TE families, while RepeatProteinMask detected TE-related sequences via homology to conserved TE protein domains. In the latter approach, RepeatModeler v1.0.836 and LTR_FINDER v1.0.637 were used to construct a de novo repeat library with candidate TE consensus sequences. This custom library was then used as a reference for RepeatMasker to annotate de novo-identified REs. Finally, the integrated annotation revealed a total of 296.37 Mb of repetitive sequences in the maroon clownfish genome, accounting for 33.51% of the assembled genome (Table 4, Fig. 3). Among these REs, DNA transposons were the most abundant component, representing 17.49% of the genome (154.69 Mb), followed by long interspersed nuclear elements (LINEs; 6.84%, 60.48 Mb) and long terminal repeats (LTRs; 3.29%, 29.08 Mb) (Table 4).
Prediction and functional annotation of protein-coding genes
Prior to protein-coding gene prediction and structural annotation, repetitive regions in the assembled genome were masked to minimize interference from repetitive sequences on gene model construction. An integrated strategy of de novo prediction, homology-based annotation, and RNA-seq-supported annotation was applied for comprehensive protein-coding gene annotation. First, AUGUSTUS v3.2.138 and GlimmerHMM v3.0.439 were employed to perform the ab inito gene structure prediction. Second, GeMoMa v1.6.440 was employed to transfer gene annotations from five evolutionarily related fish species. Second, homologous protein sequences were downloaded from the NCBI database for the following species: Amphiprion ocellaris (clown anemonefish, GCA_022539595.1), Amphiprion clarkii (yellowtail clownfish, GCA_027123335.1), Lates calcarifer (Asian seabass, GCA_051027255.1), Perca flavescens (yellow Perch, GCA_004354835.1) and Stegastes partitus (Bicolor damselfish, GCA_000690725.1). These protein sequences were aligned to the masked genome so as to guide homology-based gene structure prediction. Third, RNA-seq data derived from the four tissues (Table 1) were assembled into transcript contigs with Trinity v2.5.141 using default parameters. These assembled transcripts were then used as evidence for gene structure refinement via PASA v2.3.342. Finally, gene models generated from the three approaches were integrated using the Evidence Modeler (EVM) pipeline v1.043, with weights assigned to each evidence type (RNA-seq > homology > de novo) to prioritize reliable gene structures.
This integrated annotation yielded a total of 24,556 high-confidence protein-coding genes, with an average gene length of 19.72 kb and an average coding sequence (CDS) length of 1,734.11 bp (Table 5). Completeness of the predicted gene set was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.4.744 against the actinopterygii_odb10 database revealing that 99.12% of the complete BUSCO orthologs were successfully recovered, which confirms the high quality of gene annotation.
Functional annotation of these predicted protein-coding genes was conducted using Blastp v2.2.2645, with deduced protein sequences aligned against five publicly available databases, including NCBI Non-Redundant Protein Sequence (NR) database, Swiss-Prot46, Gene Ontology (GO)47 (for functional classification), Kyoto Encyclopedia of Genes and Genomes (KEGG)48 (for pathway annotation), and EuKaryotic Orthologous Groups (KOG)49 (for orthologous gene family classification). All alignments were performed with an E-value threshold of 1e-5 to ensure significant sequence homology. In total, 23,361 protein-coding genes (95.13% of the total predicted gene set) were successfully annotated with at least one functional hit in at least one of the five searched databases (Table 6).
Data Records
Sequencing data (including MGI, PacBio, ONT, Hi-C, and transcriptome sequencing) and the assembled genome of maroon clownfish were deposited in the National Center for Biotechnology Information (NCBI) database under the BioProject accession number PRJNA130608050. Raw sequencing reads are available in the Sequence Read Archive (SRA) with accession number SRP61780451. The genome assembly, predicted coding sequences, and functional annotation files of maroon clownfish were deposited in Figshare (No: https://doi.org/10.6084/m9.figshare.30104977)52. The complete genome assembly has also been deposited at the NCBI under accession number of GCA_053813585.153.
Technical Validation
To evaluate the quality of maroon clownfish genome assembly, four complementary approaches were employed (Table 7). First, BUSCO v5.4.744 was employed to evaluate assembly completeness against the actinopterygii_odb10 database. Our results showed that 99.98% of the complete BUSCOs were recovered, including 99.73% as single-copy complete genes (S) and 0.25% as duplicated complete genes (D), confirming high overall completeness of the assembly. Second, Merqury v1.32854, a k-mer-based quality evaluation tool, was applied to assess base-level accuracy and assembly completeness. K-mers were generated from MGI short reads and PacBio HiFi long reads, respectively. This analysis yielded a Quality Value (QV) of 44.37 (short reads) and 71.01 (long reads), indicating high base-level accuracy of the assembly. Third, Clipping information for Revealing Assembly Quality (CRAQ) v1.0955, a tool leveraging read clipping signals to quantify assembly accuracy, was used with PacBio HiFi and Illumina reads. The analysis determined a Reference-based Assembly Quality Indicator (R-AQI) of 97.93 and a Sample-based Assembly Quality Indicator (S-AQI) of 98.98, further verifying high accuracy of the assembled genome. Fourth, we mapped the sequencing data to the assembled genome using bwa v0.7.1756 and minimap2 v2.2657, which demonstrated mapping rates of 99.46% for the MGI data, 99.99% for the PacBio data, and 98.43% for the ONT data. Collectively, these quality metrics confirm that the maroon clownfish genome assembly is of high quality.
For the predicted protein-coding genes, additional quality evaluations were performed. In fact, BUSCO assessment against the actinopterygii_odb10 database showed a completeness value of 99.12% (Table 7), indicating comprehensive gene prediction. To validate annotation accuracy, transcriptome data were aligned to the assembled genome using STAR v2.7.11b58, and exonic coverage was calculated using BEDTools v2.29.259. Our results showed that 95.89% of exonic regions were covered by sequencing reads, further confirming high accuracy of the gene annotation (Table 7).
Data availability
In this study, sequencing data for P. biaculeatus, including MGI, PacBio, Nanopore, Hi-C, and transcriptome sequencing, have been deposited in the NCBI BioProject database under the accession number PRJNA130608050. The raw sequencing reads are available in the NCBI Sequence Read Archive (SRA) under the accession numbers SRP61780451. The final genome assembly and predicted coding sequences files were stored in Figshare (No: m9.figshare.30104977)52. The genome assembly has also been deposited at the NCBI under accession number of GCA_053813585.153 for public availability.
Code availability
The versions and parameters of bioinformatics tools applied in this study have been described in the Methods section. If no parameter is provided, the default is set. No custom code was used.
References
Allen, G.R. Damselfishes of the South Seas. New Jersey: Tropical Fish Hobbyist Publications 1-273 (1975).
Dixson, D. L. et al. Influence of prior residents on settlement preferences in the anemonefish, Premnas biaculeatus. Coral Reefs 37(2), 519–526 (2018).
Herrera, M. et al. Anemonefishes: A model system for evolutionary genomics. F1000Research 12, 204 (2023).
Marcionetti, A. et al. Insights into the genomics of clownfish adaptive radiation: the genomic substrate of the diversification. Genome Biology and Evolution 15(7), evad088 (2023).
Warner, R. R. Sex change and the size-advantage model. Trends in Ecology & Evolution 3(6), 133–136 (1988).
Hattori, A. et al. Sex change strategies and group structure of damselfishes. In Biology of damselfishes. CRC Press 55-83 (2016).
Munday, P. L. et al. Diversity and flexibility of sex-change strategies in animals. Trends in Ecology & Evolution 21(2), 89–95 (2006).
Mondal, R. C. et al. Beyond the Binary: Controlling Natural Sex‐Change in Hermaphroditic Fishes. Reviews in Aquaculture 17(4), e70066 (2025).
Zhang, X. et al. A telomere-to-telomere gap-free genome assembly of the protandrous hermaphrodite Asian seabass (Lates calcarifer). Scientific Data 12, 1457 (2025).
Zhang, K. et al. A telomere-to-telomere genome assembly of the protandrous hermaphrodite blackhead seabream, Acanthopagrus schlegelii. Scientific Data 12, 350 (2025).
Manabe, H. et al. Field evidence for bi‐directional sex change in the polygynous gobiid fish Trimma okinawae. Journal of Fish Biology 70(2), 600–609 (2007).
Srinivasan, M. et al. Experimental evaluation of the roles of habitat selection and interspecific competition in determining patterns of host use by two anemonefishes. Marine Ecology Progress Series 186, 283–292 (1999).
Dunn, D. F. The clownfish sea anemones: Stichodactylidae (Coelenterata: Actiniaria) and other sea anemones symbiotic with pomacentrid fishes. Transactions of the American Philosophical Society 71(1), 3–115 (1981).
Fishelson, L. Behaviour, socio‐ecology and sexuality in damselfishes (Pomacentridae). Italian Journal of Zoology 65(S1), 387–398 (1998).
Mariscal, R. N. et al. Field guide to anemonefishes and their Host Sea anemones. Copeia 1993(3), 899 (1993).
Mei, L. et al. Evaluation of QIAamp® DNA Stool Mini Kit for ecological studies of gut microbiota. Journal of Microbiological Methods 54(1), 13–20 (2003).
Chen S. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17), i884-i890.
Rhoads, A. et al. PacBio Sequencing and Its Applications. Genomics Proteomics & Bioinformatics 13, 278–289 (2015).
De Coster, W. et al. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34(15), 2666–2669 (2018).
Marçais, G. et al. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27(6), 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33(14), 2202–2204 (2017).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36(9), 2896–2898 (2020).
Langmead, B. et al. Fast gapped-read alignment with Bowtie 2. Nature Methods 9(4), 357–359 (2012).
Dekker, J. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology 16, 259 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3(1), 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101 (2016).
Xu, M. et al. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9(9), giaa094 (2020).
Xu, G. C. et al. LR_Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. Gigascience 8(1), giy157 (2019).
Takai, A. Karyotype Differentiation characterized by C-banded heterochromatin and nucleolus organizer regions in anemonefish (Pomacentridae). Cytologia 86(2), 175–179 (2021).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture Research 10(8), uhad127 (2023).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27(2), 573–580 (1999).
Tarailo-Graovac, M. et al. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics Chapter 4, 4–10 (2009).
Wang, X. & Wang, L. GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Frontiers in Plant Science 7, 1350 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Science of the United States of America 117, 9451–9457 (2020).
Xu, Z. et al. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–268 (2007).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–439 (2006).
Majoros, W. H. et al. TigrScan and GlimmerHMM: two open sources ab initio eukaryotic gene-finders. Bioinformatics 20(16), 2878–2879 (2004).
Keilwagen, J. et al. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods in Molecular Biology 1962, 161–177 (2019).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512 (2013).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31(19), 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Altschul, S. F. et al. Basic local alignment search tool. Journal of Molecular Biology 215(3), 403–410 (1990).
Bairoch, A. et al. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research 28(1), 45–48 (2000).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nature Genetics 25(1), 25–29 (2000).
Kanehisa, M. et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research 44(D1), D457–D462 (2016).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
NCBI Bioproject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1306080 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP617804 (2025).
Zhang, X. Genome assembly, predicted coding sequences and functional annotation files of P. biaculeatus. Figshare https://doi.org/10.6084/m9.figshare.30104977 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_053813585.1 (2025).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21(1), 245 (2020).
Li, K. et al. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nature Communications 14(1), 6556 (2023).
Li, H. et al. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14), 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18), 3094–3100 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1), 15–21 (2013).
Quinlan, A. R. et al. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6), 841–842 (2010).
Acknowledgements
This work was supported by Shenzhen Natural Science Foundation (no. JCYJ20241202124511016) and Shenzhen Industrial Development Special Fund Project (no. 1303).
Author information
Authors and Affiliations
Contributions
Q.S. and T.L. conceived and designed the study. X.Z., C.H., M.Y. and Y.Y. collected samples. X.Z., J.C. and C.H. performed data analysis. T.L., M.Y., Y.Y. and C.H. conducted experiments for species identification. X.Z., C.H. and T.L. wrote the manuscript. Q.S. revised the manuscript. All authors read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Hu, C., Chen, J. et al. A telomere-to-telomere gap-free genome assembly of the protandrous maroon clownfish (Premnas biaculeatus). Sci Data 13, 220 (2026). https://doi.org/10.1038/s41597-025-06538-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06538-9
