
Abstract
The longfin barb (Acrossocheilus longipinnis), a vulnerable cyprinid fish endemic to China’s Pearl River basin, is of significant conservation concern and also popular in the ornamental fish trade. To facilitate genetic research and molecular breeding for this species, we generated a high-quality genome by integrating PacBio HiFi long reads and Hi-C sequencing data. The final assembly spans approximately 936.04 Mb, achieving high continuity with a contig N50 of 36.09 Mb. Assessment of genome quality revealed excellent completeness (98.76% BUSCO score) and accuracy (QV = 54.46; GCI = 29.76; CRAQ = 96.40). The vast majority of the sequence (927.20 Mb, 99.06%) was successfully anchored to 25 chromosomes. Annotation predicted 24,718 protein-coding genes and identified approximately 553.06 Mb (59.09%) of repetitive elements. This high-quality chromosome-scale reference genome provides a crucial foundation for investigating the genomic underpinnings of A. longipinnis evolution and will significantly advance molecular breeding programs aimed at its conservation and sustainable utilization.
Similar content being viewed by others


Background & Summary
The cyprinid genus Acrossocheilus Oshima, 1919 comprises 26 valid species distributed across East and Southeast Asia, including mainland China, Taiwan, Hainan, Laos, and Vietnam. These small- to medium-sized barbines are principally characterized by a medially interrupted lower lip with two thick lateral lobes, which are anteriorly separated from the lower jaw by a distinct groove running the entire length of the jaw1. These species are widely distributed across Laos, Vietnam, and southern China, including Hainan, Taiwan, and other parts of the Chinese mainland2. Acrossocheilus longipinnis, is an endemic species of mainland China currently known only from the Pearl River basin, exhibits an elongated, laterally compressed body covered in dense scales with a prominent lateral line. Its silver-gray base coloration is adorned with five distinct pale yellow vertical bars. A key morphological trait in males is the elongation of the last branched ray and first unbranched ray of the dorsal fin into filamentous projections. Valued in the ornamental fish trade for its unique morphology and striking coloration, this species has experienced significant wild population declines, as indicated by recent fishery resource assessments. This decline is attributed to multiple anthropogenic threats, including cascading hydropower dam construction, extensive sand mining, overfishing, environmental pollution, and the introduction of invasive fish species. Consequently, A. longipinnis has been classified as Vulnerable on the IUCN Red List.
Molecular research on A. longipinnis remains limited. To date, only its mitochondrial genome has been sequenced3. Crucially, a reference genome assembly for this species is still lacking, which significantly hinders progress in understanding its biology, advancing genetic breeding programs, and developing desirable aquacultural traits. Recent advancements in DNA sequencing technologies, however, offer unprecedented opportunities for genomic research. Notably, Pacific BioSciences’ (PacBio) Circular Consensus Sequencing (CCS) mode provides long read lengths (10–20 kb) and high accuracy (>99%), thus greatly facilitating de novo assembly studies of both plant and animal genomes4,5. According to the comprehensive overview by Li and Durbin6, high-fidelity (HiFi) sequencing enables near-telomere-to-telomere assemblies by resolving repetitive regions and segmental duplications that are challenging for short-read approaches. In a parallel manner, Wang et al.7 emphasize HiFi’s applications in complex genomic regions, such as centromeres and ribosomal DNA arrays, and its superiority in variant detection and phasing compared to other long-read platforms like Oxford Nanopore Technologies7. When integrated with complementary approaches such as chromosomal conformational capture (Hi-C) sequencing, these technologies enable the generation of highly contiguous, chromosome-level genome assemblies. Such integrated approaches have already been successfully applied in another Acrossocheilus species, Acrossocheilus fasciatus, demonstrating their utility in resolving genomic architectures within this genus8.
Here, we assembled a high-quality genome of A. longipinnis by combining short sequencing reads, PacBio HiFi long reads, and Hi-C sequencing data. The final longfin barb genome assembly had a total length of 936.04 Mb, with 99.06% (927.20 Mb) of the sequences successfully anchored to 25 chromosomes. The assembly demonstrated high continuity (contig N50 = 36.09 Mb) and completeness (BUSCO = 98.76%), supported by quality metrics including a QV value of 54.46, a GCI score of 29.76, and a CRAQ value of 96.40. Subsequent annotation identified 24,718 protein-coding genes and 553.06 Mb of repetitive sequences. This high-quality genome assembly not only facilitates population genetic research and evolutionary analyses of A. longipinnis but also provides valuable resources for optimizing genetic breeding efforts.
Methods
Sampling, DNA and RNA extraction
This study was carried out according to the recommendations for the care and use of animals for scientific purposes set up by the Animal Care and Use Committee of the Chinese Academy of Fishery Sciences (ACUC-CAFS). Samples of A. longipinnis were collected from Hechi City, Guangxi Zhuang Autonomous Region, China (coordinates: 107°33′–108°13′ E, 24°22′–24°55′ N). Tissue samples were promptly collected, snap-frozen in liquid nitrogen, and then stored at −80 °C. DNA and RNA extraction, library construction, and sequencing in this study were performed using standard experimental and analytical protocols provided by NextOmics Biosciences (Wuhan, China).
Long read DNA preparation and sequencing
A total of 8 μg of high-quality genomic DNA was extracted from muscle tissue using a Qiagen DNeasy Blood and Tissue Kit (Qiagen, USA) according to the manufacturer’s instructions. The quality and concentration of the extracted DNA were assessed using a NanoDrop One spectrophotometer (Thermo Scientific, USA) and 1% agarose gel electrophoresis. PacBio long insert libraries were prepared using the SMRTbell Express Template Prep Kit 2.0 according to manufacturers’ instructions, with an insert size of approximately 20 kb. The libraries were sequenced on the PacBio Revio system in CCS mode. Subreads were processed with SMRTLink (v11.1.0)9 using the parameters “--minPasses 3 --minPredictedAccuracy 0.99 --minLength 500”, producing approximately 114.37 Gb HiFi reads with an N50 size of 16,728 (Table 1). The parameter “minPredictedAccuracy” set to 0.99 in the context of PacBio SMRTLink software means that, during the data processing of sequencing reads, only those reads that have a predicted accuracy of 99% or higher will be retained for further analysis.
Short read DNA preparation and sequencing
The extracted DNA (~5 μg) was randomly sheared into approximately 350 bp fragments, and a short fragment library was constructed using the MGIEasy Universal DNA Library Prep Set (MGI, China). Sequencing was conducted on the MGISEQ T7 platform (MGI, China), resulting in a total of 56.50 Gb of short sequencing reads, each 150 bp in length (Table 1).
Hi-C DNA library preparation and sequencing
A Hi-C library was generated using the DpnII restriction enzyme (GrandOmics, China). Muscle tissue samples were treated with 1% formaldehyde at room temperature for 10–30 minutes to crosslink chromatin-interacting proteins. Subsequently, the DNA was digested with the restriction enzyme, and the 5′ overhangs were repaired with a biotinylated residue. A paired-end library with insert sizes of approximately 300 bp was prepared and then sequenced on the MGISEQ T7 platform (MGI, China). A total of 127.92 Gb of clean data was obtained from 129.09 Gb of sequencing data using the software fastp (v0.19.5)10 with parameters “-w 16 --length_required 150” (Table 1).
RNA library preparation and sequencing
For the purpose of RNA sequencing, we extracted total RNA from muscle, heart, liver, spleen, gill, kidney, skin, and fin tissues using the TRIzol reagent (Invitrogen, USA) following the manufacturer’s protocol. Mixed total RNA purity was assessed with a NanoPhotometer spectrophotometer (IMPLEN, CA, USA), while RNA concentration was quantified using the Qubit RNA Assay Kit with a Qubit 2.0 Fluorometer (Life Technologies, CA, USA). RNA-seq libraries were prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina, USA) according to the manufacturer’s instructions. Sequencing was performed on a MGISEQ T7 platform (MGI, China), generating 150 bp paired-end reads.
Genome size estimation
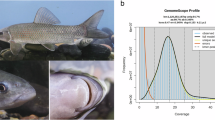
The genome size of A. longipinnis was estimated through k-mer profiling. First, raw short sequencing reads underwent quality control using fastp (v0.19.5)10. Using K-mer analysis (K = 21) of quality-filtered short reads, the genome size of A. longipinnis was first estimated with findGSE (v1.94.R)11. The genome size of A. longipinnis was estimated to be 961,326,620 bp (Fig. 1).

K-mer frequency distribution estimated. The observed K-mer (raw K-mer) frequencies (in grey), fitted K-mer frequencies (in blue) with skew normal distribution model, and overall fitting (in red) that concatenated observed and fitted K-mer frequencies.
De novo assembly and Hi-C assembly
Primary contigs were assembled from HiFi reads using Hifiasm (v 0.25.0)12 with parameters: -t 100–n-hap 2–telo-m TTAGGG hifi.fa. Genome base errors (single-nucleotide variants and small indels) were corrected using NextPolish (v1.4.1)13, integrating both HiFi reads and quality-filtered short reads. This yielded 132 contigs spanning 936.78 Mb with an N50 of 33.36 Mb. For chromosomal anchoring, BWA (v0.7.12)14 was used to align the Hi-C clean data to the assembled contigs. Low-quality reads were filtered using the HiC-Pro pipeline15 with default parameters. The remaining valid reads were employed to anchor chromosomes using Juicer16 and the 3d-dna pipeline17, followed by manual correction with Juicebox (v2.13.07)18. In the 3d-DNA pipeline, a default gap size of 500 bp was inserted between consecutive sequences. Next, we applied the LR_Gapcloser19 program to close the gaps in the assemblies. To enhance genome quality, the assemblies were polished with NextPolish2 (v0.2.0)20 using HiFi reads and quality-filtered short reads. Ultimately, 99.06% of contig sequences were anchored to 25 pseudochromosomes, with only two gaps remaining (one each in pseudochromosomes 5 and 20) (Table 2 and Fig. 2). The sizes of these two gaps were 3 bp and 151 bp, respectively. The longest and shortest pseudochromosomes measured 56.97 Mb and 28.75 Mb, respectively (Table 3). The final assembly totaled 936.04 Mb with a contig N50 of 36.09 Mb (Table 2 and Fig. 3).

Hi-C assembly of chromosome interactive heat map. The abscissa and ordinate represent the order of each bin on the corresponding chromosome group. The colour block illuminates the intensity of interaction from white (low) to red (high).

Snail plot showing the features of the assembled A. longipinnis genome. The main plot is divided into 1,000 size-ordered bins around the circumference with each bin representing 0.1% of the 936,040,231 bp assembly. The distribution of chromosome lengths is shown in dark grey with the plot radius scaled to the longest chromosome present in the assembly. Orange and pale-orange arcs show the N50 and N90 chromosome lengths (36,094,363 and 29,100,020 bp), respectively. The pale grey spiral shows the cumulative chromosome count on a log scale with white scale lines showing successive orders of magnitude. The blue and pale-blue area around the outside of the plot shows the distribution of GC, AT and N percentages in the same bins as the inner plot.
Repetitive sequence annotation
Repeat elements in the A. longipinnis genome were annotated employing a combined methods of homology alignment and de novo searches. The homology-based blast was performed against the RepBase database (http://www.girinst.org/repbase/)21 using RepeatMasker (v4.0.7)22 and Proteinmask software for known repeat elements. For de novo annotation, we firstly employed LTR_FINDER (v1.06)23 and RepeatModeler (v1.0.4)24 to bulid a de novo repeat library, and then was used to predict repeat elements using RepeatMasker (v4.0.7)22 with default parameters. Additionally, Tandem Repeat Finder (v4.10.0)25 was used to discern tandem repeats with default parameters. In detail, a total of 553.06 Mb (~59.09%) of repetitive sequences were obtained. Among the interspersed repeats, long terminal repeats were the most prevalent type, accounting for 32.67% of the genome (Table 4).
Gene prediction and functional annotation
Gene prediction was performed using a multifaceted approach incorporating transcriptome-based, homology-based, and ab initio methods. For the transcriptome-based prediction, a total of 8.73 Gb of RNA-seq clean reads were aligned to the A. longipinnis assembly using Hisat2 (v2.2.1)26 (Table 5). Stringtie (v1.2.2)27 was then utilized to assemble transcripts based on the alignment results. In addition, the RNA-seq data were de novo assembled by Trinity (v2.15.2)28 with parametrs:–seqType fq–max_memory 200 G–min_kmer_cov 2–min_glue 2–CPU 60–min_contig_length 200. Afterwards, the assembled transcripts were aligned against the A. longipinnis assembly using Program to Assemble Spliced Alignment (PASA; v2.4.1)29. For homology-based prediction, we utilized Miniport (v0.11) to conduct a comparative analysis of the protein sequences from seven vertebrate species, including A. fasciatus8, Ctenopharyngodon idella30, Cyprinus carpio31, Poropuntius huangchuchieni32, Onychostoma macrolepis (GCF_012432095.1), Danio rerio (GCF_049306965.1), and Homo sapiens (GCF_009914755.1). For ab initio prediction, 2,000 high-quality genes from PASA were randomly selected as the training set for model training with AUGUSTUS (v3.2.3)33. AUGUSTUS (v3.2.3)33 was then employed to predict coding regions in the repeat-masked genome. In addition, Fgenesh (v2.4.5)34 was also used for ab initio prediction. Finally, all gene models were integrated using EvidenceModeler (v2.1.0)35. The final comprehensive gene set comprised 24,718 genes (Table 6), with an average of 10.44 exons per gene, an exon length of 170.64 bp, and a coding sequence (CDS) length of 1781.09 bp.
After gene prediction, the finalized gene sets derived from the preceding methods underwent functional annotation through matching with a variety of databases. Briefly, amino-acid sequences were aligned to SwissProt36, Kyoto Encyclopedia of Genes and Genomes (KEGG)37, and the NCBI nonredundant database (NR) using the Diamond (v 2.1.10)38 with an E-value cutoff of 1e-05. Protein domains were identified using the InterProScan (v5.30)39 program, and Gene Ontology (GO) terms for each gene were also extracted through InterProScan. Overall, 24,228 (98.02%) of the predicted protein-coding genes were functionally annotated (Table 6).
Ethical approval
The study did not involve any wild animals. All experimental procedures involving fish were conducted in strict compliance with the Guide for the Hongshui River Rare Fish Conservation Center to minimize animal suffering and ensure animal welfare.
Data Records
The raw sequencing data have been deposited into the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with accession number SRP60447140 under BioProject number PRJNA1297891. Additionally, the genome assembly and annotation are available at the Figshare dataset41.
Technical Validation
Genome assembly and gene prediction quality assessment
We employed a multi-faceted approach to rigorously evaluate the precision and integrity of the A. longipinnis genome assembly. First, we utilized Merqury (v1.3)42 with a combination of HiFi long reads and short reads, setting the K-mer value at 19, to calculate the consensus QV. The analysis yielded a QV of 54.46, indicating a high level of accuracy in the assembled genome sequence (Table 2). Subsequently, we aligned the HiFi reads and quality-filtered short reads to the assembly using minimap2 (v2.24-r1122)43 and BWA (v0.7.12)14, respectively. This process demonstrated an exceptional alignment rate, with 99.99% of the HiFi reads and 99.85% of the short sequencing reads successfully mapped to the genome (Table 2). Centromeric regions were predicted following the method described in the recent telomere-to-telomere genome study of Cyprinus carpio31. We found the centromeric regions displayed the canonical features of centromeres: high repetitive sequence content, low gene density, and low HiFi read coverage depth, aligning with the previous research reports31,44 (Fig. 4). Additionally, both assembly gaps were located within highly repetitive regions, one of which lay within a centromere. The HiFi read coverage in the regions flanking these gaps was notably lower compared to the genome-wide average. Clipping information for revealing assembly quality (CRAQ, v1.10)45 was used to assess the accuracy of our genome assembly based on PacBio HiFi and quality-filtered short reads, resulting in a S-AQI of 96.40, confirming high assembly quality. In addition, genome continuity inspector (GCI, v1.0)46 yielded a value of 29.76, which was comparable to that of the chicken complete genome47. To assess genome completeness, we performed an analysis with Benchmarking Universal Single-Copy Orthologs (BUSCO) (v5.5.0)48 using the actinopterygii_odb10 database. The results showed that 98.76% of the BUSCO genes were complete, including 97.53% single-copy and 1.24% duplicated orthologs, while only 0.93% of the genes were fragmented (Fig. 5). Furthermore, BUSCO analysis of the genome annotation revealed 97.14% of the recognized BUSCOs were complete, consisting of 95.11% single-copy and 2.03% duplicated genes (Fig. 5). Collectively, these comprehensive evaluation metrics strongly suggest that the A. longipinnis genome assembly has achieved a high standard of quality, providing a reliable resource for subsequent genetic and biological studies.

Characterization of centromeric regions and gap locations visualized by a circos plot. From inside to outside: Gene density in 1 Mb sliding windows; Percentage of repetitive sequence in 1 Mb sliding windows; Centromere density in 1 Mb sliding windows; Gap locations; HiFi reads coverage depth; The length of pseudochromosome in the size of Mb.

BUSCO assessments of A. longipinnis genome and gene sets.
Data availability
Raw sequencing data have been deposited in the NCBI SRA database under BioProject accession number PRJNA1297891, with accession numbers as follows: PacBio HiFi: SRR3477099149; Hi-C: SRR3477099250; RNA sequencing: SRR3477099051; DNA short-read sequencing: SRR3477099352. The genome assembly has been uploaded to the GenBank database under the accession GCA_054083375.153. Moreover, the genome assembly, annotation files (GFF3, FASTA), and gene functional annotation datasets, are available via Figshare41. All datasets are publicly accessible without restrictions.
Code availability
No specific code or script was used in this work. Commands used for data processing were all executed according to the manuals and protocols of the corresponding software.
References
Yuan, L. Y., Liu, X. X. & Zhang, E. Mitochondrial phylogeny of Chinese barred species of the cyprinid genus Acrossocheilus Oshima, 1919 (Teleostei: Cypriniformes) and its taxonomic implications. Zootaxa 4059, 151–168 (2015).
Chen, T. E. et al. A New Species of the Genus Acrossocheilus Oshima, 1919 (Cypriniformes: Cyprinidae) from the Dabie Mountains. Animals 15, 734 (2025).
Hou, X.-J. et al. Complete mitochondrial genome of the freshwater fish Acrossocheilus longipinnis (Teleostei: Cyprinidae): genome characterization and phylogenetic analysis. Biologia 75, 1871–1880 (2020).
Wenger, A. M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature biotechnology 37, 1155–1162 (2019).
Lovell, J. T. et al. Four chromosome scale genomes and a pan-genome annotation to accelerate pecan tree breeding. Nature Communications 12, 4125 (2021).
Li, H. & Durbin, R. Genome assembly in the telomere-to-telomere era. Nature Reviews Genetics 25, 658–670 (2024).
Wang, B. et al. Long and Accurate: How HiFi Sequencing is Transforming Genomics. Genomics Proteomics Bioinformatics 23 (2025).
Zheng, J. et al. Chromosome-level genome assembly of Acrossocheilus fasciatus using PacBio sequencing and Hi-C technology. Scientific Data 11, 166 (2024).
Chin, C. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods 10, 563–569 (2013).
Chen, S., Zhou, Y., Chen, Y. & Jia, G. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Sun, H., Ding, J., Piednoël, M. & Schneeberger, K. findGSE: estimating genome size variation within human and Arabidopsis using k-mer frequencies. Bioinformatics (Oxford, England) 34, 550–557 (2018).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics (Oxford, England) 36, 2253–2255 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Servant, N. et al. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biology 16 (2015).
Durand, N. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, eaal3327 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell systems 3, 99–101 (2016).
Xu, G.-C. et al. LR_Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. GigaScience 8 (2018).
Hu, J. et al. NextPolish2:a repeat-aware polishing tool for genomes assembled using HiFi long reads. (bioRxiv, 2023).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467 (2005).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics (Oxford, England) 21(Suppl 1), i351–8 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–8 (2007).
Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 5 (2004).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biology 20, 278 (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644–652 (2011).
Haas, B. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31, 5654–5666 (2003).
Liu, F. et al. The telomere-to-telomere gapless genome of grass carp provides insights for genetic improvement. GigaScience 14 (2025).
Yuan, J. et al. A telomere-to-telomere genome assembly of koi carp (Cyprinus carpio) using long reads and Hi-C technology. GigaScience 14 (2025).
Chen, L. et al. Chromosome-level genome of Poropuntius huangchuchieni provides a diploid progenitor-like reference genome for the allotetraploid Cyprinus carpio. Molecular ecology resources 21, 1658–1669 (2021).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic acids research 33, W465–7 (2005).
Solovyev, V., Kosarev, P., Seledsov, I. & Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome biology 7(Suppl 1), S10.1–12 (2006).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Research 27, 49–54 (1999).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 28, 27–30 (2000).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60 (2015).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP604471 (2025).
Li, J. Chromosome-level genome assembly of Acrossocheilus longipinnis using PacBio sequencing and Hi-C technology. Figshare. Dataset. https://doi.org/10.6084/m9.figshare.29665907.v1 (2025).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Yin, D. et al. Telomere-to-telomere gap-free genome assembly of the endangered Yangtze finless porpoise and East Asian finless porpoise. GigaScience 13 (2024).
Li, K., Xu, P., Wang, J., Yi, X. & Jiao, Y. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nature Communications 14, 6556 (2023).
Chen, Q., Yang, C., Zhang, G. & Wu, D. GCI: a continuity inspector for complete genome assembly. Bioinformatics 40 (2024).
Huang, Z. A.-O. et al. Evolutionary analysis of a complete chicken genome. Proc Natl Acad Sci USA. 120(8), e2216641120 (2023).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
NCBI sequence read archive https://identifiers.org/ncbi/insdc.sra:SRR34770991 (2025).
NCBI sequence read archive https://identifiers.org/ncbi/insdc.sra:SRR34770992 (2025).
NCBI sequence read archive https://identifiers.org/ncbi/insdc.sra:SRR34770990 (2025).
NCBI sequence read archive https://identifiers.org/ncbi/insdc.sra:SRR34770993 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_054083375.1 (2025).
Acknowledgements
This work is supported by Operating funds of Hongshui River Rare Fish conservation Center.
Author information
Authors and Affiliations
Contributions
Zechen E conceived this study, designed the experiment, and performed data analysis. Fangyuan Xiong contributed to the experimental design, collected samples, and performed data analysis. Yuansheng Zhu and Li Wang provided funding and contributed to conceptualization. Jiajun Zhang and Shenghui Dong assisted in methodology and data curation. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
E, Z., Xiong, F., Zhu, Y. et al. Chromosome-level genome assembly of the longfin barb (Acrossocheilus longipinnis). Sci Data 13, 600 (2026). https://doi.org/10.1038/s41597-026-06656-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06656-y
