
Abstract
The casuarina moth, Lymantria xylina, is a serious pest threatening subtropical regions through severe defoliation and strong invasive potential. Despite its economic impact and high invasion risk, a high-quality reference genome remains lacking. To bridge this knowledge gap, we generated a chromosome-level genome assembly for L. xylina combining Illumina short-reads, Oxford Nanopore long-reads, and high-throughput chromatin conformation capture (Hi-C) scaffolding data. Following long-reads based assembly and Hi-C scaffolding, the final genome assembly totals 977.74 Mb, with 930.50 Mb (95.17%) of sequences anchored onto 31 pseudo-chromosomes, achieving a scaffold N50 of 34.15 Mb. The genome assembly, featuring fully assembled telomeres on all 31 pseudo-chromosomes, demonstrates 94.5% Benchmarking Universal Single-Copy Orthologs (BUSCO) completeness and high accuracy with consensus quality value of 31.72. Repetitive elements constitute 77.18% of the genome, and 18,484 protein-coding genes were predicted, with 95.21% functionally annotated. This high-quality genome assembly provides a critical foundation for elucidating interaction mechanisms with host plants and natural enemies (nucleopolyhedrovirus, Beauveria bassiana), for developing enhanced pest management and control strategies.
Similar content being viewed by others
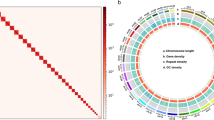
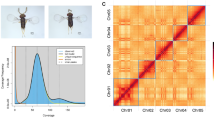

Data availability
The raw sequencing reads have been deposited in GSA (CRA027397, https://ngdc.cncb.ac.cn/gsa). Additionally, raw high-throughput sequencing data for L. xylina have been deposited in the NCBI Sequence Read Archive with accession number SRP655858. The genome assembly of L. xylina is available in GWH (GWHGEMT00000000.1, https://ngdc.cncb.ac.cn/gwh) and GenBank (JBPSJU000000000). Genome annotation and related data are available at Figshare (https://doi.org/10.6084/m9.figshare.29497898). All untargeted metabolomic data used in this publication have been deposited to the EMBL-EBl MetaboLights database with the identifier MTBLS13575 (https://www.ebi.ac.uk/metabolights/MTBLS13575). Additionally, the processed metabolomics dataset, including metabolite identification tables and quality control metrics, has been deposited in Figshare at https://doi.org/10.6084/m9.figshare.30909260.
Code availability
No custom code was used for this study. All data analyses were performed using published bioinformatics software with specified parameter settings, as stated in the Methods section and Figshare65.
References
Wallner, W. E. & McManus, K. A. Proceedings, Lymantriidae: A comparison of features of New and Old World tussock moths. Gen. Tech. Rep. NE-123. Broomall, PA: US Department of Agriculture, Forest Service, Northeastern Forest Experiment Station. 554 p. 123, 1-554 (1989).
Shen, T.-C., Tseng, C.-M., Guan, L.-C. & Hwang, S.-Y. Performance of Lymantria xylina (Lepidoptera: Lymantriidae) on artificial and host plant diets. J Econ Entomol 99, 714–721 (2006).
Pogue, M. A review of selected species of Lymantria Hübner (1819)(Lepidoptera: Noctuidae: Lymantriinae) from subtropical and temperate regions of Asia, including the descriptions of three new species, some potentially invasive to North America. (U.S. Department of Agriculture, Forest Health Technology Enterprise Team, 2007).
Hwang, F. Investigation of the larval food plant range of the casuarina moth, Lymantria xylina. (Doctoral dissertation, MS thesis, National Chung Hsing University. Taichung, Taiwan, 2005).
Hwang, S.-Y., Hwang, F.-C. & Shen, T.-C. Shifts in developmental diet breadth of Lymantria xylina (Lepidoptera: Lymantriidae). J Econ Entomol 100, 1166–1172 (2007).
Hwang, S.-Y., Jeng, C.-C., Shen, T.-C., Shae, Y.-S. & Liu, C.-S. Diapause termination in casuarinas moth (Lymantria xylina) eggs. Formos. Entomol 24, 43–52 (2004).
Chang, Y. & Weng, Y. Morphology, life habit, outbreak and control of casuarina tussock moth (Lymantria xylina Swinhoe). Quart J Chin Forest 18, 29–36 (1985).
Zhang, J. et al. Reproductive and flight characteristics of Lymantria xylina (Lepidoptera: Erebidae) in Fuzhou, China. Insects 15, 894 (2024).
Mastro, V. C., Munson, A. S., Wang, B., Freyman, T. & Humble, L. M. History of the Asian Lymantria species program: A unique pathway risk mitigation strategy. J Integr Pest Manag 12, 31 (2021).
Zhang, J. et al. Evaluation of the potential flight ability of the casuarina moth, Lymantria xylina (Lepidoptera: Erebidae). Insects 15, 506 (2024).
Nai, Y.-S. et al. Genomic sequencing and analyses of Lymantria xylina multiple nucleopolyhedrovirus. BMC genomics 11, 116 (2010).
Tsay, J.-G., Lee, M.-J. & Chen, R.-S. Evaluation of Beauveria bassiana for controlling casuarina tussock moth (Lymantria xylina Swinhoe) in casuarina plantations. Taiwan Journal for Science 16, 201–207 (2001).
Cheng, T. et al. Genomic adaptation to polyphagy and insecticides in a major East Asian noctuid pest. Nat Ecol Evol 1, 1747–1756 (2017).
Xie, Q. et al. Identification of differentially expressed genes and proteins related to diapause in Lymantria dispar: Insights for the mechanism of diapause from transcriptome and proteome analyses. PloS one 20, e0316065 (2025).
Blackburn, G. S. et al. Genetics of flight in spongy moths (Lymantria dispar ssp.): functionally integrated profiling of a complex invasive trait. BMC genomics 25, 541 (2024).
Akhanaev, Y. et al. Virulence and genome analysis of baculovirus isolates from different Lymantria dispar populations. Sci Rep 15, 28449 (2025).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884-–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432 (2020).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736 (2017).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963 (2014).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259 (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–1125 (2013).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic Res 10, uhad127 (2023).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–W268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Hoede, C. et al. PASTEC: an automatic transposable element classification tool. PloS one 9, e91929 (2014).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110, 462–467 (2005).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 5, 4.10.1–4.10.14 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res 44, e89–e89 (2016).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, 215–225 (2003).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Alioto, T., Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr Protoc Bioinformatics 64, e56 (2018).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268, 78–94 (1997).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 21 (2004).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, e78–e78 (2015).
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M. & Buell, C. R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC genomics 7, 327 (2006).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
McGinnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32, W20–W25 (2004).
Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol 5, R7 (2004).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–370 (2003).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18, 366–368 (2021).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–D124 (2005).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014).
Chong, J. & Xia, J. MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 34, 4313–4314 (2018).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genom Proteom Bioinf 19, 578–583 (2021).
Database resources of the national genomics data center, China national center for bioinformation in 2024. Nucleic Acids Res 52, D18-D32 (2024).
NGDC/CNCB. Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/search?searchTerm=CRA027397 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP655858 (2025).
NGDC/CNCB. Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/98230/show (2025).
Wang, R. et al. Chromosome-level genome assembly and genome annotation of Lymantria xylina. GenBank http://identifiers.org/ncbi/insdc:JBPSJU000000000 (2025).
Wang, R. et al. Chromosome-level genome assembly and genome annotation of Lymantria xylina. Figshare https://doi.org/10.6084/m9.figshare.29497898 (2025).
Wang, R. MetaboLights MTBLS13575 http://www.ebi.ac.uk/metabolights/MTBLS13575 (2025).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol 20, 277 (2019).
Xu, Z. et al. Chromosome-level genome assembly of the Asian spongy moths Lymantria dispar asiatica. Sci Data 10, 898 (2023).
Wang, R. et al. Commands and parameters settings. Figshare https://doi.org/10.6084/m9.figshare.30909005 (2025).
Acknowledgements
This research was funded by the National Natural Science Foundation of China (grant numbers: 31600522) and the Natural Science Foundation of Fujian Province (grant number: 2017J0106).
Author information
Authors and Affiliations
Contributions
W.R. designed this study. S.L., H.J., T.N. carried out genome sequencing. S.L., H.J., T.N., K.W., X.H., S.W. performed the data analyses and visualization. S.L., H.J., T.N. drafted the manuscript. W.R., F.Z. revised the manuscript. All authors contributed the final text of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, S., Jiang, H., Ni, T. et al. Chromosome-level genome assembly of the casuarina moth, Lymantria xylina Swinhoe (1903). Sci Data (2026). https://doi.org/10.1038/s41597-026-06724-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-026-06724-3
