
Abstract
The hadal trench is the deepest part of the global ocean and harbors highly abundant microbial cells. However, the diversity and function of the majority of microbial communities in this part of the ocean are still unclear. Here, we collected 35 metagenomes from three push cores across different sites in both the northern and southern Yap trench to construct a comprehensive gene and genome dataset. A total of 32 million non-redundant genes were predicted from the whole metagenome datasets, with 63% assigned to known functional groups based on currently available databases. A total of 404 metagenome-assembled genomes (MAGs) with completeness >50% and contamination <10% were retrieved, and their taxonomy was highly diverse across 26 phyla. Alpha- and Gammaproteobacteria, Phycisphaerae, Nitrospiria, and Dehalococcoidia were dominant classes across all samples. The nonredundant gene and MAG datasets are valuable resources for advancing our understanding of the diversity, composition, and functions of microbiota in the sediment of the hadal trench.
Similar content being viewed by others
Background & Summary
The hadal zone is the deepest habitat of the ocean, referring to the deep region with >6000 meters water depth, and approximately 1%–2% of the global benthic area, but constitutes the deepest 45% of the vertical depth gradient1. Tectonically, the hadal zone is in the subduction zone, creating topographic V-shaped depressions that form a unique topographic feature in the deep ocean2,3. The geophysical and geochemical features of the hadal zone are distinctive from those of other habitats in the deep ocean1,4. Topography, geographical isolation, and spatio-temporal variation in food supply, as well as low temperature and extremely high hydrostatic pressure, created a unique habitat that accommodated a diverse and active microbial community4. With advances in deep-sea sampling technologies and in high-throughput sequencing, the knowledge of the hadal biosphere has been largely improved. Sediments of the hadal zone harbor microbial communities with high abundance and diverse metabolic functions, showing clear shifts of composition and assembly strategies from bathyal and abyssal sediments to deep hadal zones5,6,7,8,9. The proportion of heterotrophic microbial communities was dominant in hadal sediments10,11. They are able to degrade various organic matter, such as aromatic compounds, alkane, and long-chain hydrocarbons, as revealed from previous metagenome sequencing-based analyses5,6,8,12. Further, growing evidence indicates that chemoautotrophic carbon fixation occurred within the hadal trench13,14. Despite the increasing number of studies in the hadal biosphere, the current microbiome data are not sufficient to carry out a comprehensive investigation on microbial diversity and function in the sediment of hadal trenches. Therefore, the knowledge of the diversity, composition, and function of microbial communities in the hadal sediment remains deficient.
The Yap Trench is located at the southern end of the Philippine Sea Plate and is a tectonic region of convergence among the Philippine Sea, the Pacific and the Caroline plates in the southwestern of the Pacific Ocean. There are three different trenches, namely the Yap Trench, the Mariana Trench and the Palau Trench, created by the process of tectonic plate collisions15. The Yap Trench is located between the Mariana Trench and the Palau Trench, extending about 700 km long and 50 km wide from the trench axis to the island arc. The width of the Yap Trench is much less than that of other arc-trench systems, forming a sharp “V” shape. The Yap Trench is divided into northern and southern sections, with the boundary between them marked at 8°26′N, based on its relation to the Caroline Ridge16,17. The geological, geophysical, and geochemical characteristics were different between northern and southern sections16,17. For instance, the southern Yap Trench has a gentler trench slope and lower seismic intensity compared to the northern section18. Additionally, the concentration of organic matter in the sediment of the southern section is higher than that of the northern section19,20. These contrasting characteristics may influence the formation of different microbial communities in the sediments of the two sections of the Yap Trench, which is important to broad our understanding of microbial community functions on hadal zone.
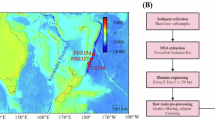
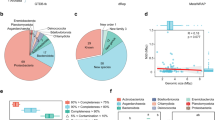
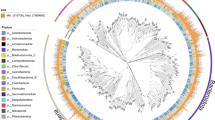
To better understand the diversity, composition and function of sediment microbial communities at the Yap Trench, and compare the microbiome of northern and southern parts, we collected three push cores from different water depths, covering abyssal (Sites 1 and 2) and hadal trench (Site 3) regions in the northern and southern parts of the Yap Trench. 35 metagenomes obtained from top to bottom layers of three push cores (Fig. 1A and Supplementary Table S1). Through metagenome assembly and binning processes (Fig. 1B), we obtained 32 million non-redundant predicted genes and 404 metagenome assembled genomes (MAGs) with completeness >50% and contamination <10% from the whole dataset. Within these MAGs, 142 MAGs were estimated to be >70% completeness, account for 35% of total MAGs (Supplementary Table S2). Based on taxonomy classification and the relative abundance of these MAGs, Alpha- and Gammaproteobacteria, Phycisphaerae, Nitrospiria and Dehalococcoidia were dominant classes across all samples. Gammaproteobacteria and Acidimicrobiia were highly abundant in abyssal sediment, while Alphaproteobacteria and Dehalococcoidia were dominant classes in the hadal sediments (Fig. 2). The assembled contigs of each sample were integrated and redundant genes were removed with sequence similarity (cut off = 99%, Fig. 1B). After clustering, 3,976,582 non-redundant genes were retrieved from the datasets. We blasted these non-redundant genes against the KEGG, Pfam, CAZy, and eggNOG databases to predict their functions, and 63% of these genes could be assigned to known genes in the databases (Fig. 3). The MAGs with more than 70% completeness and less than 10% contamination were used to construct a phylogenomic tree (Fig. 4). The results showed that the taxonomy of MAGs included 26 phyla indicating highly diverse of microbial community in Yap trench sediment (Fig. 4). Among them, top three numbers of MAGs were affiliated with Pseudomonadota (n = 50), Acidobacteriota (n = 17) and Chloroflexota (n = 8). The archaeal MAGs belonged to Thermoproteota and Nanoarchaeota (Supplementary Table S2). These datasets will enable us to further understand the diversity, composition and function of microbiota in the hadal trench, and highlight their critical roles in the hadal biosphere.

The map of sampling sites and pipeline of metagenome analysis (A). Sampling location. The red dots showed the location of sampling sites (B). The pipeline of metagenomic analysis for sediment samples.

The relative abundance and distribution of recovered MAGs in three sites. The stacked bar plot based on the relative abundance of MAGs obtained from this study.

Functional characterization of the non-redundant gene catalog. Non-annotation indicates that these genes were not annotated in at least one of the following databases: eggNOG, Pfam, KEGG, and CAZy. The low panel of filled-in cells indicated the databases are in an intersection. The vertical bars in top panel represent the number of annotated genes in the intersection or shared between different databases in intersection. The horizontal bars in left panel indicate the total number of annotated genes in each database.

Phylogenetic tree of MAGs including bacteria and archaea. The tree was constructed based on concatenated 37 conserved single copy proteins alignment. The black points in the branches of the tree represent bootstrap values >0.7. The MAGs recovered from this study were labeled red. Phyla are color-coded, and taxonomy is from the Genome Taxonomy Database (GTDB). The gray bars in the outside cycle indicated the relative abundance of MAGs.
Methods
Sample collection
Three push cores were retrieved from the western trench slope of Yap Trench during R/V Xiangyanghong 10th cruise with manned submersible Jiaolong (Fig. 1). The subsample of sediment was split at 1-cm intervals using sterilized tools on board; additionally, the subsamples were split at 2-cm intervals below 10 cm. Only the interior sections of the sediment were used for microbiological study to avoid potential contamination21. A total of 35 subsamples obtained from 3 push cores were analyzed (Supplementary Table S1), and subsequently, sediments for microbiological analyses were stored at −80 °C until further processing.
DNA extraction and sequencing
Total DNA was extracted from the sediments with the PowerSoil DNA Isolation Kit (Qiagen, Germany) according to the manufacturer’s instructions. The DNA was purified and concentrated with the Genomic DNA Clean & Concentrator kit (Zymo Research, USA). DNA was fragmented into smaller pieces with a Covaris instrument (Covaris, USA) and selected 300–500 bp DNA fragments to construct libraries with Illumina Nextera DNA libraries kit (Illumina, USA), and sequencing on Illumina HiSeq X-Ten platform (Wuhan Onemore-tech Co., Ltd.).
Metagenome assembly and binning
The trimming of raw reads was performed using Trimmomatic v.0.39. The clean reads of each sample were assembled using MEGAHIT v1.2.9 with parameters ‘–k-min 21–k-max 144–k-step 10’22. The length of contigs larger than 1000 bp was used for downstream analysis. The coverage of contigs was determined using BWA software (v0.7.17; BWA-MEM algorithm)23. Binning process performed with metaWRAP binning module (v1.3.2; parameters: -metabat2, -maxbin2, -concoct, -m 2000)24 and VAMB25 with default parameters, respectively. The reconstructed MAGs were refined using the ‘bin_refinement’ module of MetaWRAP v1.312, and their quality and taxonomic information were identified using CheckM2 v1.0.226 and GTDB-TK v2.4.027 with the GTDB-TK reference database (version 220), respectively. MAGs with completeness more than 50% and contamination less than 10% were used for downstream analysis. 404 representative MAGs were obtained based on an average nucleotide identity (ANI) cutoff value of 95% with dRep v3.5.028. The coverage of each MAG was calculated using CoverM in genome mode (v0.6.1; https://github.com/wwood/CoverM; parameters: -min-read-percent-identity 0.95, -min-read-aligned-percent 0.75, -trim-min 0.10, -trim-max 0.90, -m relative_abundance).
Functional gene annotation and phylogenetic analysis
The open reading frames (ORFs) of the genomes and contigs were predicted using Prodigal v2.6.329 with the ‘-p meta’ parameter and then annotated against the Kyoto Encyclopedia of Genes and Genomes (KEGG) (version Jan. 1st, 2025) using KofamScan v1.3.030 with E-values ≤ 1e-20, and Tigrfam31 using hmmscan (v3.3.2)32. The peptidase and proteinase encoding genes were annotated in the MEROPS database 12.433 using Diamond blastp v0.9.1434 with a threshold of coverage >40% and E-value < 1e-20.
We used 142 MAGs with completeness >70% and contamination <10% to construct the phylogenetic tree. The concatenated set of 37 conversed single-copy genes based on a hidden Markov Model profile was used for phylogenetic analysis with IQ-TREE (v2.2.0.3)35 with the best-fit model (Q.pfam + I + I + R9) and 1000 times ultrafast bootstrapping. The tree file was edited using the online tool iTOL (https://itol.embl.de/).
Data Records
The 35 raw metagenome sequences are available on the NCBl Sequence Read Archive (SRA) associated with BioProject number PRJNA131417336 and accession number SRP61789737. A total of 404 non-redundant FASTA formatted MAGs from these metagenomes were available at European Nucleotide Archive (ENA) under accession code PRJEB10696838, PRJEB10696939 and PRJEB10691440. The detailed information for these qualified MAGs, including genomic quality, GTDB taxonomy, accession number and relative abundance was shown in the Supplementary Table S2.
Technical Validation
To avoid contamination of sediment samples, all sampling tools and containers have been sterilized before sampling and only the interior sections of the sediment core were collected for DNA extraction. After the samples collection, the sediment samples were stored at −80 °C until further processing. All processes of DNA extraction and library construction were carried out in an ultra-clean lab. To ensure the quality of genes prediction, we selected assembled contigs with a length larger than 1000 bp. To maximize the number of MAGs, the length of contigs more than 1000 bp and four different binning tools were used in the binning process, such as CONCOCT, MetaBat2, Maxbin2 and VAMB. The quality of MAGs was identified with CheckM2. The high-quality MAGs were completeness >50% and contamination <10%. To increase the accuracy of phylogenetic analysis, we used MAGs with completeness >70% and contamination <10% to construct a phylogenetic tree.
Usage Notes
The biosphere in hadal zone sediments has many enigmas and is only partially explored. This study provides comprehensive metagenomic data from the sediments retrieved from different depths of the northern and southern Yap Trench, covering abyssal and trench sediments. The datasets contained 21 and 14 metagenomes from abyssal and trench sediments, respectively. All data were analyzed with a commonly used pipeline, generating bacterial and archaeal high-quality MAGs. The datasets can be used for exploring the diversity and potential metabolic function of microorganisms inhibited in the hadal sediment and comparing with the microbiome of other hadal trench.
Data availability
Metagenome sequences are deposited on the NCBl Sequence Read Archive (SRA) associated with BioProject number PRJNA131417336 and accession number SRP61789737. 404 high-quality non-redundant MAGs retrieved from these metagenomes of three cores were deposited in ENA database under accession code PRJEB10696838, PRJEB10696939 and PRJEB10691440, respectively. The metadata of MAGs and custom codes were available at FigShare41.
Code availability
The custom scripts to generate the datasets are publicly available on Figshare41. The parameters and versions of all bioinformatics tools used for the metagenomic analysis are described in the Methods section.
References
Jamieson, A. J., Fujii, T., Mayor, D. J., Solan, M. & Priede, I. G. Hadal trenches: the ecology of the deepest places on Earth. Trends in Ecology & Evolution 25(3), 190–7 (2010).
Stewart, H. A. & Jamieson, A. J. Habitat heterogeneity of hadal trenches: Considerations and implications for future studies. Progress in Oceanography 161, 47–65 (2018).
Stern, R. J. Subduction zones. Reviews of Geophysics 40(4), 3-1–3-38 (2002).
Du, M. et al. Geology, environment, and life in the deepest part of the world’s oceans. The Innovation 2(2), 100109 (2021).
Zhou, Y.-L., Mara, P., Cui, G.-J., Edgcomb, V. P. & Wang, Y. Microbiomes in the Challenger Deep slope and bottom-axis sediments. Nature Communications 13(1), 1515 (2022).
Liu, R. et al. Novel Chloroflexi genomes from the deepest ocean reveal metabolic strategies for the adaptation to deep-sea habitats. Microbiome 10(1), 75 (2022).
Li, Y., Cao, W., Wang, Y. & Ma, Q. Microbial diversity in the sediments of the southern Mariana Trench. Journal of Oceanology and Limnology 37(3), 1024–9 (2019).
Xiao, X. et al. Microbial ecosystems and ecological driving forces in the deepest ocean sediments. Cell 188(5), 1363–77.e9 (2025).
Fu, L. et al. Characteristics of the archaeal and bacterial communities in core sediments from Southern Yap Trench via in situ sampling by the manned submersible Jiaolong. Science of The Total Environment 703, 134884 (2020).
Peoples, L. M. et al. Microbial Community Diversity Within Sediments from Two Geographically Separated Hadal Trenches. Front Microbiol 10, 2019 (2019).
Hiraoka, S. et al. Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments. ISME J 14(3), 740–56 (2020).
Wang, Y. et al. Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ Microbiol 21(2), 716–29 (2019).
Wenzhöfer, F. et al. Benthic carbon mineralization in hadal trenches: Assessment by in situ O2 microprofile measurements. Deep Sea Research Part I: Oceanographic Research Papers 116, 276–86 (2016).
Luo, M., Gieskes, J., Chen, L., Shi, X. & Chen, D. Provenances, distribution, and accumulation of organic matter in the southern Mariana Trench rim and slope: Implication for carbon cycle and burial in hadal trenches. Marine Geology 386, 98–106 (2017).
Yang, Y. et al. Geology of the Yap Trench: new observations from a transect near 10°N from manned submersible Jiaolong. International Geology Review 60(16), 1941–53 (2018).
Xia, C.-L. et al. Geological and geophysical differences between the north and south sections of the Yap trench-arc system and their relationship with Caroline Ridge subduction. Geological Journal 55(12), 7775–89 (2020).
Fujiwara, T. et al. Morphology and tectonics of the Yap Trench. Marine Geophysical Researches 21(1), 69–86 (2000).
Jamieson, A. The Hadal Zone: Life in the Deepest Oceans. Cambridge: Cambridge University Press (2015).
Li, D. et al. Spatial heterogeneity of organic carbon cycling in sediments of the northern Yap Trench: Implications for organic carbon burial. Mar Chem 223, 103813 (2020).
Li, D. et al. Comparison of sedimentary organic carbon loading in the Yap Trench and other marine environments. Journal of Oceanology and Limnology 38(3), 619–33 (2020).
Lever, M. A. et al. Life under extreme energy limitation: a synthesis of laboratory- and field-based investigations. FEMS Microbiology Reviews (2015).
Li, D., Liu, C.-M., Luo, R., Sadakane, K., Lam, T.- W. J. B. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. 31(10):1674-6 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357 (2012).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6(1), 158 (2018).
Nissen, J. N. et al. Improved metagenome binning and assembly using deep variational autoencoders. Nature Biotechnology 39(5), 555–60 (2021).
Chklovski, A., Parks, D. H., Woodcroft, B. J. & Tyson, G. W. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nature Methods 20(8), 1203–12 (2023).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36(6), 1925–7 (2019).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 11(12), 2864–8 (2017).
Hyatt, D. et al. LJJBb. Prodigal: prokaryotic gene recognition and translation initiation site identification. 11(1):119 (2010).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36(7), 2251–2 (2019).
Haft, D. H. et al. TIGRFAMs and Genome Properties in 2013. Nucleic Acids Research 41(D1), D387–D95 (2012).
Eddy, S. R. Accelerated Profile HMM Searches. PLOS Computational Biology 7(10), e1002195 (2011).
Rawlings, N. D. & Bateman, A. How to use the database and website to help understand peptidase specificity. Protein Science 30(1), 83–92 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59 (2014).
Minh, B. Q. et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 37(5), 1530–4 (2020).
NCBI BioProject https://identifiers.org/ncbi/bioproject:PRJNA1314173 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP617897 (2025).
Euuropean Nucleotide Archive https://www.ebi.ac.uk/ena/browser/view/PRJEB106968 (2026).
Euuropean Nucleotide Archive https://www.ebi.ac.uk/ena/browser/view/PRJEB106969 (2026).
Euuropean Nucleotide Archive https://www.ebi.ac.uk/ena/browser/view/PRJEB106914 (2026).
Mingyang, N. et al. 35 metagenomic datasets from the northern and southern slope of Yap trench sediments. Figshare https://doi.org/10.6084/m9.figshare.29328314 (2025).
Acknowledgements
This work was supported by National Natural Science Foundation of China (42030407), National Key Basic Research and Development Project of China (2015CB755904), National Natural Science Foundation of China (42006083, 41906124), Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (SML2023SP220, Dong, SML2024SP002).
Author information
Authors and Affiliations
Contributions
M.N. and L.F.: designed this study, performed data analysis, interpreted the data and wrote manuscript. D.L. collected samples. D.L., Q.Y., M.W., C.L. and Z.H. edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Niu, M., Fu, L., Yan, Q. et al. 35 metagenomic datasets from the northern and southern parts of the Yap trench sediments. Sci Data 13, 422 (2026). https://doi.org/10.1038/s41597-026-06812-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06812-4
