
Abstract
Mitochondrial DNA (mtDNA) mutations are critical to disease research, evolutionary studies, and lineage tracing but are challenging to analyze due to interference from nuclear mitochondrial sequences (NUMTs). Current high-throughput sequencing techniques rely on multiple primers or probes to amplify short mtDNA fragments, followed by alignment to a reference genome. However, this approach fails to mitigate NUMTs interference, leading to ambiguous results. In this study, we presented a nanopore-based third-generation sequencing (TGS) method using a single primer pair to amplify full-length mtDNA, effectively circumventing NUMTs artifacts. Sequencing was carried out on the QITAN TECH QNome-3841hex platform, generating complete mtDNA coverage for 106 samples from eight distinct family pedigrees, including complex familial structures such as half-siblings and multi-generational households. The sequencing achieved 100% genome coverage with an average mapping rate of 99.96%, supporting comprehensive genome characterization. The resulting dataset offers valuable insights into mtDNA mutation detection, mitochondrial genetics, population genetics, ancestry tracing, and forensic identification, and may advance mtDNA sequencing technologies and intergenerational studies.
Similar content being viewed by others


Background and summary
Mitochondria are organelles responsible for energy production and play key roles in cellular functions such as biosynthesis, signaling, and regulating apoptosis1,2. Mutations in mitochondrial DNA (mtDNA) can disrupt these processes and lead to human diseases and mitochondrial disorders3,4,5. Unlike nuclear DNA (nDNA), paternal mtDNA is typically eliminated after fertilization6, making mtDNA inheritance strictly maternal. This maternal inheritance pattern allows mtDNA to reflect key population genetic characteristics, such as migration history, population structure, and adaptive evolution7,8,9. Furthermore, mtDNA’s double-stranded, circular structure and its multiple copies make it more resistant to environmental degradation than nDNA. This resilience makes mtDNA a useful marker for analyzing highly degraded, or low-template DNA samples, especially in forensic science. Comprehensive studies of mtDNA variation are therefore common in fields such as genetic diversity, tracing maternal lineage, and investigating the role of mitochondrial mutations in disease.
Current mtDNA analysis predominantly relies on Sanger sequencing or next-generation sequencing (NGS). These methods have advanced forensic identification, human evolution research, population genetics and disease pathogenesis10,11,12. However, both techniques have limitations. Sanger sequencing suffers from low throughput and reduced sensitivity in detecting low-frequency mutations, while NGS faces challenges such as short read lengths and GC bias13,14,15,16. In conventional NGS-based mtDNA sequencing, read lengths are typically limited to 100–150 bp, making haplotype resolution challenging in mixed samples.
To address these limitations, long-read sequencing (LRS) platforms have recently been introduced. These platforms generate reads that span the entire mitochondrial genome, thereby eliminating alignment gaps at the junctions of the circular genome and enabling accurate haplotype phasing17. This capability enables accurate reconstruction of complete mitochondrial haplotypes and facilitates the detection of large structural variants, such as long deletions or rearrangements, that are often missed by NGS.
However, practical challenges remain. PCR-based enrichment methods may introduce coverage bias, and distinguishing true mitochondrial sequences from nuclear mitochondrial DNA segments (NUMTs) continues to be a significant concern18. NUMTs originate from ancient insertions of mitochondrial DNA into the nuclear genome and are distributed across nearly all chromosomes. Crucially, recent genomic benchmarks highlight that NUMT insertion is an ongoing process, generating polymorphic NUMTs across human populations19. In parallel, the utilization of more comprehensive reference genomes (e.g., T2T-CHM13), in contrast to earlier reference-based surveys, has revealed a substantially expanded catalog of structurally resolved NUMTs20. Certain regions, including chromosomes 2, 8, and X, contain particularly dense NUMT clusters, with insertion lengths ranging from a few hundred base pairs to more than 10 kb21 (Fig. 1). Notably, “mega-NUMTs” exceeding 5 kb in size, remain problematic even with long-read sequencing, particularly in regions sharing high sequence homology (e.g., the well-characterized hotspots on chromosomes 1 and 17)22. Distinguishing these large insertions from genuine mtDNA molecules continues to present technical challenges, thereby interfering with variant calling and haplotype interpretation23.

NUMTs distribution across chromosomes and lengths. (a) Number of NUMTs per chromosome based on the UCSC Genome Browser (hg38, NuMTs Sequence track). (b) Length of NUMTs based on NuMTs Sequence track. NUMTs smaller than 500 bp are shown in the inset.
Although challenges remain, TGS represents the most practical and informative approach currently available for comprehensive mtDNA characterization. Its raw error rate (typically 5–10%) and the persistence of mega-NUMT interference preclude absolute sequence accuracy, yet the ability to generate full-length, single-molecule reads provides a robust foundation for reliable variant detection and haplotype phasing when combined with stringent quality control and filtering. In particular, when TGS is integrated with targeted long-range PCR strategies that preferentially amplify circular mitochondrial templates, it enables dependable differentiation between authentic mtDNA and NUMT-derived sequences. This combination offers a balanced framework that emphasizes data completeness and analytical reproducibility rather than maximal per-base resolution.
While advances in sequencing technologies have improved mitochondrial genomics, there remains a substantial gap in mtDNA research concerning multi-generational pedigrees and complex familial structures. Limited publicly available datasets hinder comprehensive assessment of maternal inheritance stability and intergenerational mutation dynamics. Expanding high-quality, full-length mtDNA sequencing resources in familial contexts is therefore essential for elucidating mitochondrial transmission patterns and improving the reliability of mtDNA-based genetic studies. To address this need, we generated full-length mtDNA sequences from 106 individuals across eight family pedigrees using the QITAN QNome-3841hex nanopore sequencing platform. While initial basecalling was performed by the sequencing provider, all subsequent data processing and analysis were conducted using a transparent and reproducible bioinformatics pipeline. The workflow includes quality evaluation, alignment performance assessment, verification of maternal lineage consistency through familial checks, and evaluation of potential interference from NUMTs. Together, these datasets provide a rigorously validated and well-documented resource for investigating maternal inheritance and mutation dynamics, serving as a valuable benchmark for future mtDNA research in long-read sequencing contexts.
Methods
Ethical statement
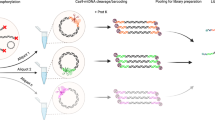
The study was conducted in accordance with the ethical standards of Fudan University and the 1964 Helsinki Declaration, along with its later amendments. All samples were collected following the approval of the Ethics Committee of Fudan University (No. 2022-11). Informed consent was obtained from all participants, and written consent was provided prior to their participation in the study (Fig. 2). For participants under the age of 18 or those unable to provide informed consent independently, written consent was obtained from their legal guardians or authorized representatives. The Ethics Committee explicitly granted a consent waiver for the processing and sharing of fully anonymized genetic data under Approval No. 2022-11, as no personally identifiable information was retained in the dataset. All samples and sequencing records were irreversibly de-identified before analysis, ensuring compliance with ethical standards for participant privacy protection.

Full-length Mitochondrial Genome Sequencing using third-generation sequencing technology.
Sample collection and DNA extraction
Peripheral blood samples were collected from eight pedigrees, with a total of 106 individuals participating. Blood was collected in BD Vacutainer® tubes (BD, USA). Genomic DNA was extracted using the QIAamp DNA Blood Kit (QIAGEN, Netherlands), following the manufacturer’s protocol. DNA concentration was measured using a Qubit® 4.0 fluorometer (Thermo Fisher Scientific, USA), and the extracted DNA was stored at –80 °C for future use. Each individual was assigned a unique identifier, which is consistently used across sample processing, sequencing, and analysis. These identifiers, together with pedigree information, demographic variables, parental identifiers, and SRA accession numbers, are summarized in the sample metadata file (available at Figshare24).
Library construction and sequencing
Library preparation was carried out using 3 ng DNA and a Full-Length Mitochondrial DNA Identify kit (QITAN TECH, China). A single pair of primers (universal amplification primer, UAP) targeting the conserved regions of the mitochondrial DNA was designed to amplify the entire 16,569 bp mtDNA fragment. While the exact primer sequences are not publicly disclosed by the manufacturer, both primers are located within the 16S rRNA gene region, around position 2,900 of the mitochondrial genome. The primer positions are indicated in Fig. 3. First-round PCR was carried out in a 25 μL reaction mixture containing 12.5 μL of QITAN Long Fragment Amplification (LPA) Mix, 2.5 μL of UAP, 3 ng of genomic DNA, and nuclease-free water (NFW) to adjust the final volume. Amplification was performed using the PCR Thermal Cycler (Hangzhou LongGene Scientific Instruments, China) with an initial denaturation at 98 °C for 2 min, followed by 10 cycles of 98 °C for 10 s, 64 °C for 10 s, and 68 °C for 3 min 30 s, then 20 cycles of 98 °C for 10 s and 68 °C for 3 min 30 s, with a final extension at 68 °C for 5 min. PCR products were analyzed by agarose gel electrophoresis to confirm the presence of a primary band around 16 kb.

Primer positions within the human mitochondrial genome. Schematic circular map of the mitochondrial genome showing annotated functional regions. Primer binding sites are indicated at ~2.9 kb within the 16S rRNA region.
First-round PCR products were directly used for a second round of amplification without purification. The second-round PCR was performed in a 25 μL reaction containing 1–2 μL of first-round PCR product, 12.5 μL of LPA Mix, 2 μL of barcode primer, and NFW to reach the final volume. The cycling conditions included an initial denaturation at 98 °C for 2 min, followed by 8 cycles of 98 °C for 10 s and 68 °C for 3 min 30 s, with a final extension at 68 °C for 5 min. The amplified products were purified using 2 μL of purification enhancer (BPR) and 15 μL of VAHTS® DNA Clean Beads (1:0.6 ratio, Vazyme, China) to remove residual primers and PCR components.
Following amplification and purification, the amplicons underwent end repair and adapter ligation according to the manufacturer’s protocol. For end repair, 0.15–0.3 pmol of purified PCR product was mixed with 7 μL of DNA Repair Buffer (DRB), 3 μL of End-Prep Mix (EPM), and NFW up to 60 μL, followed by incubation at 20 °C for 10 min, 65 °C for 10 min, and hold at 12 °C. The reactions were purified using 60 μL of VAHTS DNA Clean Beads (1:1 ratio) and eluted with 70 μL of NFW. Adapter ligation was performed by adding 5 μL of Sequencing Adapter Complex (SAC), 25 μL of 4 × Ligation Reaction Buffer (LRB), and 10 μL of DNA Ligase Enzyme (DLE) to the purified DNA, followed by incubation at room temperature for 10 min. The final libraries were purified using 40 μL of VAHTS DNA Clean Beads (1:1 ratio) and eluted in 22 μL of AEB Elution Buffer. The libraries were then quantified and normalized to 5–7 fmol before sequencing on the QNome-3841hex system using the QCell-3841hex chip (QITAN TECH, China).
Quality control and data analysis
Raw sequencing signal data was processed by the sequencing service provider (QITAN TECH, China) to generate initial FASTQ files. This procedure included basecalling using the provider’s proprietary model version 3.1.3 (QDSv1.0_QCell384v1.0_QDLEvB20_NTGv3.1.3) and an initial quality filtering step to remove reads with a mean quality score below Q7. The resulting FASTQ files, which serve as the input for the reproducible analysis pipeline detailed below, have been deposited in a public repository (see Data Records).
To prepare the data for variant analysis, reads were filtered by length to retain sequences between 5,000 and 18,000 bp, corresponding to the expected amplicon size generated by the long-range PCR strategy targeting the entire ~16.6 kb mitochondrial genome. Length filtering and per-read statistics—including number of reads, total bases, read length mean and median, N50, GC content, base-weighted mean Q and mean error rate were computed using a custom Python script. Quality metrics for the filtered reads were further assessed using FastQC (version 0.12.1)25, and summary reports were aggregated with MultiQC (version 1.28)26.
Filtered reads were aligned to the revised Cambridge Reference Sequence (rCRS; NC_012920.1)27 using Minimap2 (version 2.28)28 with the options -ax map-ont and–secondary = no. The resulting BAM files were processed using Samtools (version 1.21)29 for sorting and indexing, and analyzed using the mtDNA-Server 2 (version 2.1.16)30 in local mode. Within the mtDNA-Server 2 pipeline, contamination detection was performed using Haplocheck (version 1.3.3)31, and haplogroup classification was carried out using HaploGrep 3 (version 3.2.2)32 based on Phylotree Build 1733. A variant allele frequency (VAF) threshold of 10% was applied to generate a high-confidence set of single nucleotide variants and short indels. The variant calls were output in a standard Variant Call Format (VCF). Consensus sequences for each sample were generated by applying the variants detected from the BAM alignments (AF ≥ 0.1) to the rCRS reference sequence using Bcftools (version 1.22)29.
To generate data for the assessment of potential NUMT contamination, the length-filtered reads were also aligned to the human reference genome hg3834 (with chrM replaced by rCRS), with multi-mapping alignments retained. This process identified reads aligning to both mitochondrial and nuclear loci.
Structural variant detection was performed using Sniffles2 (version 2.3.2)35 on filtered and sorted BAM files. Only variants ≥ 50 bp with a minimum read support of 5 were considered, and variants with an allele fraction below 8% were excluded to account for potential sequencing errors.
Multiple sequence alignment of all 106 consensus sequences was performed with MAFFT (version 7.505)36. Phylogenetic reconstruction was carried out with IQ-TREE (version 2.0.7)37 under the GTR + G substitution model with 1000 ultrafast bootstrap replicates. The resulting phylogenetic tree was visualized using FigTree (version 1.4.4)38.
Data Records
The raw sequencing data files (FastQ format) for the full-length mtDNA data generated by QNome-3841hex nanopore sequencer have been deposited to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA). The data can be accessed under SRA accession number SRP57037539. Each sample is identified using a unique individual ID, which corresponds directly to the filenames of the uploaded FastQ files. The consensus mitochondrial genome sequences have been submitted to GenBank and are accessible via BioProject accession number PRJNA123594740. The variant data for this study are accessible via the European Variation Archive (EVA) at EMBL-EBI under accession number PRJNA123594741.
Additionally, comprehensive metadata, quality control metrics, and analysis results are available on Figshare with the https://doi.org/10.6084/m9.figshare.3085656824. This repository includes the Sample Metadata file (Data Record 1), which provides detailed demographic and lineage information for each sample. Sequencing performance is summarized in the Sequencing Metrics and Alignment Stats files (Data Records 2 and 3), covering yield, quality scores, and mapping rates. The identification of nuclear mitochondrial DNA segments is detailed in the NUMT Analysis file (Data Record 4). Furthermore, the repository contains the Full Pedigree Dataset (Data Record 5) illustrating the maternal relationships for all eight families, and the aggregated MultiQC Reports (Data Record 6) for data quality assessment.
Raw signal files are currently unavailable from the vendor. Should they become available in the future, we will ensure deposition to enable re-basecalling and further improvement of data accuracy. This dataset provides resources for comparative studies, population genetics, and family pedigree analysis. Researchers are encouraged to utilize these data for additional investigations, validations, or meta-analyses.
Technical Validation
To ensure the reliability and accuracy of the results, multiple technical validation measures were implemented throughout the experiment.
Quality control of library preparation
Strict quality control measures were applied throughout the study to monitor the reliability of the sequencing workflow. Positive control DNA (9947 A) and negative control (NFW) were included in each experimental run to monitor the reliability of the procedure. The positive control confirmed successful amplification and sequencing, while the negative control checked for potential contamination. Control results were closely monitored, and only experiments meeting the expected performance criteria were deemed valid for downstream analysis. The integrity of full-length mtDNA amplification was further evaluated by agarose gel electrophoresis. Representative results from 24 randomly selected samples out of the 106 individuals are shown in Fig. 4, each band corresponding to a separate individual. The consistent presence of dominant ~16 kb bands indicated successful amplification of the full-length mitochondrial genome.

Technical validation of full-length mitochondrial DNA amplification. Agarose gel electrophoresis results of 24 representative samples randomly selected from the 106 individuals are shown. Each lane corresponds to one independent individual.
Sequencing read quality and alignment assessment
Sequencing read quality was evaluated using standard metrics, including read length distribution, N50, GC content, and per-base quality scores. Quality metrics for the length-filtered FASTQ files were generated using FastQC and NanoPlot, with summary reports aggregated by MultiQC (Fig. 5). Summary statistics for all samples are provided in the sequencing metrics file (available at Figshare24). Across samples, the mean per-base Phred quality score ranged between 13.36 and 17.14, corresponding to estimated error rates of ~7.67–12.44%.

Sequence Quality Histogram. Per-base Phred quality scores across all samples after quality filtering. The complete MultiQC report is available on Figshare24 (Data Record 6).
To further validate the data’s utility, the filtered reads were aligned to the rCRS reference. The alignments demonstrated consistently high mapping rates (mean 99.97%), complete mitochondrial genome coverage (100%), and high, uniform read depth (mean 1,896.46×). These metrics confirm that the sequencing data comprehensively covers the entire mitochondrial genome for all samples (see sequencing metrics file).
Genetic consistency across familial lineages
The pedigree structure of this dataset allows internal validation of the sequencing and variant-calling results, providing a means to evaluate its reliability for lineage-based studies. To perform this validation, variants and haplogroups were determined using mtDNA-Server 2. Maternal inheritance patterns were then assessed across the 106 sequenced samples. Among these, 74 samples belong to 20 distinct maternal lineages, while the remaining 32 samples have no maternal relationship within the dataset (e.g., spouses or father–child pairs) and were therefore excluded from mother–offspring concordance evaluation.
Within the 74 maternally related samples, 73 (98.6%) showed consistent haplogroup assignments within the same maternal lineage (see sample metadata file). One notable exception was sample 212. As illustrated in Fig. 6, its haplogroup assignment did not match its recorded maternal lineage, suggesting a possible metadata or sampling annotation issue. The variant data for this sample have been deposited in the EVA, and its haplogroup information is summarized in the sample metadata file. The complete pedigree charts for all eight families are provided in the full pedigree dataset (available at Figshare24).

Representative pedigree (Pedigree 4) illustrating maternal inheritance of mtDNA haplotypes. Squares and circles indicate males and females, respectively. Underlined text below each symbol shows the haplogroup assigned by mtDNA-Server 2. Sample 212 exhibits a haplogroup inconsistent with its recorded maternal lineage (highlighted in red).
Phylogenetic analysis of the 106 consensus sequences (Fig. 7) also showed robust clustering of samples according to their known maternal lineages, confirming maternal consistency and supporting the reliability of the sequencing data.

Phylogenetic tree of 106 consensus mitochondrial sequences.
Structural and NUMTs assessment
Structural variants and potential contamination from nuclear mitochondrial DNA segments (NUMTs) were evaluated to confirm the dataset’s integrity. Analysis with Sniffles2 revealed no structural variants ≥ 50 bp across any sample, consistent with the expected stability of the human mitochondrial genome. Subsequent evaluation of NUMTs contamination was based on reads that aligned to both mitochondrial and nuclear loci. The vast majority of reads mapped exclusively to the mitochondrial genome, with mapping rates ≥99.07% across all samples (see the alignment stats file, available at Figshare24).
Reads that mapped to both mitochondrial and nuclear loci with high confidence (mapping quality >30) were identified across 36 samples, totaling 78 events (see the NUMT analysis file, available at Figshare24). Many of these reads overlapped with known NUMT regions in the nuclear genome, typically aligning over a long span to the nuclear locus and over shorter, fragmented regions within the mitochondrial genome. The overall pattern supports the high specificity of the sequencing and alignment workflow. Combined with the prior application of stringent length filtering and alignment parameters excluding secondary mappings (secondary = no), these results demonstrate that the dataset effectively minimizes NUMT interference and ensures high mitochondrial sequence purity.
Usage Notes
This dataset can be reused in a variety of research contexts, including forensic mtDNA genotyping, maternal lineage tracing, and population genetics. The inclusion of full-length mtDNA sequences from eight pedigrees allows users to investigate haplotype inheritance across generations, evaluate family-based variation, and explore human evolutionary patterns.
Users should be aware of the following technical considerations. Because basecalling and primary QC were performed with the provider’s proprietary and raw signals were not provided, community-developed ONT tools for re-basecalling and signal-level correction (e.g., Guppy, Bonito, Remora, Medaka) cannot be applied. Consequently, error reduction is limited to downstream alignment and variant-calling filters. To facilitate reproducibility, we provide fully specified minimap2 commands, filtering thresholds, and variant callsets (mtDNA-Server-2), allowing users to balance sensitivity against artefacts.
The provided VCF files contain a comprehensive set of variant calls generated by mtDNA-Server 2. For high-confidence analyses, we recommend using the primary variant set filtered at a 10% variant allele frequency, which has been validated for high accuracy through familial concordance checks. The VCF files report numerous low-frequency variants that may represent heteroplasmic sites. Although such variants could potentially be of biological interest (e.g., reflecting mosaicism), distinguishing true low-level heteroplasmies from platform-specific sequencing artifacts would require additional orthogonal validation. Therefore, users interested in low-level heteroplasmy are advised to interpret these sites with caution.
Finally, while our validation demonstrates that NUMT interference is minimal in this dataset due to the long-range PCR design and a 5–18 kb read length filter, users should remain mindful of important technical limitations. As noted in Background and summary, the existence of exceptionally long “mega-NUMTs” means their potential influence cannot be fully excluded, even with long-read sequencing22. Users should also note that while the single-amplicon strategy effectively captures large deletions such as the common 4.9 kb deletion, amplification bias may still occur if a structural variant spans the primer binding sites (located within the 16S rRNA region). In such instances, the mutated template may fail to amplify. Moreover, the dataset’s per-base error rate (~7.67–12.44%) overlaps with the estimated error range that can arise from NUMT-related interference42. This overlap can create ambiguity for low-frequency variants. Therefore, we strongly advise users to interpret variant calls conservatively, particularly in genomic regions known to share homology with NUMTs.
Data availability
All raw sequencing data generated in this study are available in the NCBI Sequence Read Archive (SRA) under accession SRP57037539. Consensus mitochondrial genome sequences are available in GenBank via BioProject PRJNA123594740. Variant data are available in the European Variation Archive (EVA) under PRJNA123594741. Additional supporting data, including sample metadata, pedigree charts, and quality control metrics, are available in Figshare repository24.
Code availability
All analyses were performed using publicly available tools, and no modifications were made to the original software. To ensure reproducibility, all commands and scripts used for FASTQ-based quality control, alignment and variant calling have been consolidated into a reproducible workflow. This workflow is available at https://github.com/myjasminum/mito under an open-source license.
References
Borcherding, N. & Brestoff, J. R. The power and potential of mitochondria transfer. Nature 623, 283–291, https://doi.org/10.1038/s41586-023-06537-z (2023).
Hu, Z. et al. A novel protein CYTB-187AA encoded by the mitochondrial gene CYTB modulates mammalian early development. Cell Metabolism 36, 1586–1597.e7, https://doi.org/10.1016/j.cmet.2024.04.012 (2024).
Ng, Y. S. et al. Mitochondrial disease in adults: Recent advances and future promise. The Lancet Neurology 20, 573–584, https://doi.org/10.1016/S1474-4422(21)00098-3 (2021).
Ng, Y. S. & Turnbull, D. M. Mitochondrial disease: Genetics and management. J Neurol 263, 179–191, https://doi.org/10.1007/s00415-015-7884-3 (2016).
Castellani, C. A. et al. Mitochondrial DNA copy number can influence mortality and cardiovascular disease via methylation of nuclear DNA CpGs. Genome Med 12, 84, https://doi.org/10.1186/s13073-020-00778-7 (2020).
Zhang, H., Zhu, Y. & Xue, D. Moderate embryonic delay of paternal mitochondrial elimination impairs mating and cognition and alters behaviors of adult animals. Sci. Adv. 10, eadp8351, https://doi.org/10.1126/sciadv.adp8351 (2024).
Kristjansson, D., Bohlin, J., Jugessur, A. & Schurr, T. G. Matrilineal diversity and population history of norwegians. American J Phys Anthropol 176, 120–133, https://doi.org/10.1002/ajpa.24345 (2021).
Yue, W. et al. Investigation of control region sequences of mtDNA in naqu tibetan population from northwestern China. Annals of Human Biology 48, 70–77, https://doi.org/10.1080/03014460.2021.1877351 (2021).
James, J. E., Piganeau, G. & Eyre‐Walker, A. The rate of adaptive evolution in animal mitochondria. Molecular Ecology 25, 67–78, https://doi.org/10.1111/mec.13475 (2016).
Faccinetto, C. et al. Internal validation and improvement of mitochondrial genome sequencing using the precision ID mtDNA whole genome panel. Int J Legal Med 135, 2295–2306, https://doi.org/10.1007/s00414-021-02686-w (2021).
Kopinski, P. K., Singh, L. N., Zhang, S., Lott, M. T. & Wallace, D. C. Mitochondrial DNA variation and cancer. Nat Rev Cancer 21, 431–445, https://doi.org/10.1038/s41568-021-00358-w (2021).
Parakatselaki, M.-E. & Ladoukakis, E. D. mtDNA heteroplasmy: Origin, detection, significance, and evolutionary consequences. Life 11, 633, https://doi.org/10.3390/life11070633 (2021).
Zaragoza, M. V., Fass, J., Diegoli, M., Lin, D. & Arbustini, E. Mitochondrial DNA variant discovery and evaluation in human cardiomyopathies through next-generation sequencing. PLoS ONE 5, e12295, https://doi.org/10.1371/journal.pone.0012295 (2010).
Wong, L.-J. C. Diagnostic challenges of mitochondrial DNA disorders. Mitochondrion 7, 45–52, https://doi.org/10.1016/j.mito.2006.11.025 (2007).
Li, M. et al. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. The American Journal of Human Genetics 87, 237–249, https://doi.org/10.1016/j.ajhg.2010.07.014 (2010).
Guo, Y. et al. The effect of strand bias in illumina short-read sequencing data. BMC Genomics 13, 666, https://doi.org/10.1186/1471-2164-13-666 (2012).
Shaw, J., Boucher, C., Yu, Y. W., Noyes, N. & Li, H. Long-read reconstruction of many diverse haplotypes with devider. Genome Res gr. 280510.125, https://doi.org/10.1101/gr.280510.125 (2025).
Macken, W. L. et al. Enhanced mitochondrial genome analysis: Bioinformatic and long-read sequencing advances and their diagnostic implications. Expert Rev Mol Diagn 23, 797–814, https://doi.org/10.1080/14737159.2023.2241365 (2023).
Dayama, G., Emery, S. B., Kidd, J. M. & Mills, R. E. The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res 42, 12640–12649, https://doi.org/10.1093/nar/gku1038 (2014).
Tao, Y., He, C., Lin, D., Gu, Z. & Pu, W. Comprehensive identification of mitochondrial pseudogenes (NUMTs) in the human telomere-to-telomere reference genome. Genes (Basel) 14, 2092, https://doi.org/10.3390/genes14112092 (2023).
Single-cell mitochondrial DNA sequencing: Methodologies and applications. Mitochondrial Communications 2, 107–113, https://doi.org/10.1016/j.mitoco.2024.10.001 (2024).
Wei, W. et al. Nuclear-embedded mitochondrial DNA sequences in 66,083 human genomes. Nature 611, 105–114, https://doi.org/10.1038/s41586-022-05288-7 (2022).
Xue, L., Moreira, J. D., Smith, K. K. & Fetterman, J. L. The mighty NUMT: Mitochondrial DNA flexing its code in the nuclear genome. Biomolecules 13, 753, https://doi.org/10.3390/biom13050753 (2023).
Liu, Y. A full-length mtDNA dataset for studying genetic variations across generations and complex family structures. figshare https://doi.org/10.6084/m9.figshare.30856568 (2025).
Andrews, S. A Quality Control Tool for High Throughput Sequence Data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048, https://doi.org/10.1093/bioinformatics/btw354 (2016).
Andrews, R. M. et al. Reanalysis and revision of the cambridge reference sequence for human mitochondrial DNA. Nat Genet 23, 147–147, https://doi.org/10.1038/13779 (1999).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
Weissensteiner, H., Forer, L., Kronenberg, F. & Schönherr, S. mtDNA-server 2: Advancing mitochondrial DNA analysis through highly parallelized data processing and interactive analytics. Nucleic Acids Res 52, W102–W107, https://doi.org/10.1093/nar/gkae296 (2024).
Weissensteiner, H. et al. Contamination detection in sequencing studies using the mitochondrial phylogeny. Genome Res. 31, 309–316, https://doi.org/10.1101/gr.256545.119 (2021).
Schönherr, S., Weissensteiner, H., Kronenberg, F. & Forer, L. Haplogrep 3 - an interactive haplogroup classification and analysis platform. Nucleic Acids Res 51, W263–W268, https://doi.org/10.1093/nar/gkad284 (2023).
van Oven, M. & Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 30, E386–394, https://doi.org/10.1002/humu.20921 (2009).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27, 849–864, https://doi.org/10.1101/gr.213611.116 (2017).
Smolka, M. et al. Detection of mosaic and population-level structural variants with Sniffles2. Nat Biotechnol 42, 1571–1580, https://doi.org/10.1038/s41587-023-02024-y (2024).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37, 1530–1534, https://doi.org/10.1093/molbev/msaa015 (2020).
Rambaut, A. FigTree. https://tree.bio.ed.ac.uk/software/figtree/.
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP570375 (2025).
NCBI BioProject https://identifiers.org/ncbi/bioproject:PRJNA1235947 (2025).
EMBL-EBI EVA https://www.ebi.ac.uk/eva/?eva-study=PRJNA1235947 (2025).
Gupta, R. et al. Nuclear genetic control of mtDNA copy number and heteroplasmy in humans. Nature 620, 839–848, https://doi.org/10.1038/s41586-023-06426-5 (2023).
Acknowledgements
This study was supported by National Natural Science Fund of China (82302124), National Key R&D Program of China (2024YFC3306702) and Scientific Research Projects of Science and Technology Commission of Shanghai Municipality (24JG0500500).
Author information
Authors and Affiliations
Contributions
S.Z. and A.C., conception and design. Y.X., J.Z., experiment execution. Y.N. and Q.Y., data analysis. The manuscript was written by Y.N. and Q.Y. and revised by S.Z. and A.C. All authors contributed to review the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Yang, Q., Xuan, Y. et al. A full-length mtDNA dataset for studying genetic variations across generations and complex family structures. Sci Data 13, 442 (2026). https://doi.org/10.1038/s41597-026-06824-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06824-0
