
Abstract
The striped fruit fly Zeugodacus scutellata (Diptera: Tephritidae) is a major cucurbit pest causing over 70% yield losses during outbreaks and poses an ongoing quarantine threat across Asia. To date, the absence of a high‐quality reference genome has hindered studies of its invasion dynamics and host adaptations. Here, we present a chromosome‐level assembly generated using Illumina short reads, PacBio HiFi long reads, and Hi‐C scaffolding. The final assembly is highly contiguous (contig N50 > 10.3 Kb; scaffold N50 > 75.5 Mb) and exhibits >99.3% BUSCO completeness. Annotation identified 13,327 protein‐coding genes. We also catalogued repetitive elements, providing insight into genome architecture. All raw sequencing data, assembly files, and annotations have been deposited in public repositories. This resource will enable comparative genomics of Tephritid pests, facilitate population‐level analyses of dispersal and adaptation, and support development of molecular tools for monitoring and targeted management of Z. scutellata.
Similar content being viewed by others

Background & Summary
The striped fruit fly, Zeugodacus scutellata (Diptera: Tephritidae), is one of the most destructive pests of Cucurbitaceae plants1. The species can be morphologically distinguished from other Zeugodacus spp. by the presence of three postsutural yellow vittae with a pair of medium-sized circular black spots across the face, two pairs of scutellar bristles, and the costal band slightly enlarged at the apex2. This fly species shows two annual peaks in adult abundance and survives the winter in the adult stage under leaf litter near its habitat3,4,5. Its larvae infest flowers of pumpkin and cause >70% damage during an outbreak periods, posing a serious threat to crop production6. In addition, Z. scutellata can also damage flowers, fruits and stems of other host plants, such as squash, Chinese cucumber, cucurbit, wild gourds, eggplant and pear, thus causing agricultural losses7,8. This pest is also an important quarantine species, mainly distributed in Asian countries including China, Japan, Korea, India, Thailand, Bhutan and Malaysia9,10. In 1923, Z. scutellata was first reported in Jiangsu Province of China, and currently it has been found in many regions of Southern, Central and Southwest China11. Field monitoring indicated that the abundance of this species increased in Southern of China12.
In recent years, whole-genome sequencing of fruit fly species has greatly advanced our understanding of their evolution, ecology, and pest biology. High-quality reference genomes are now available for several economically important tephritid species, such as Bactrocera dorsalis13 and Bactrocera tau14, as well as for multiple model Drosophila species. These genomic resources have enabled detailed investigations into gene family evolution, host adaptation, chemosensory perception, detoxification pathways, insecticide resistance, and population structure, and have provided essential foundations for comparative genomics and pest management strategies. However, despite its economic importance and expanding geographic distribution, genomic resources for Z. scutellata remain lacking, which has limited systematic studies of its genetic basis of host use, environmental adaptation, and population dynamics. Whole-genome sequencing of Z. scutellata is therefore necessary to fill this critical gap. A chromosome-scale reference genome provides a comprehensive framework for accurate gene prediction and functional annotation, resolves repetitive and heterozygous genomic regions, and enables robust comparative analyses with other tephritid fruit flies. Such a resource is essential for identifying genes and pathways associated with key biological traits, including chemosensation, development, reproduction, detoxification, and stress response, and for supporting population genomic studies relevant to quarantine monitoring and pest control. The genome presented in this study thus represents an important addition to fruit fly genomic resources and provides a valuable foundation for future evolutionary, ecological, and applied research on Z. scutellata.
Methods
Sample preparation
The striped fruit fly samples of Z. scutellata were collected from luffa fields in Shuangjin Village, Jiangnan Subdistrict, Yongkang City, Zhejiang Province (28.884365°N, 120.008902°E) on July 12 and August 27, 2024, respectively. After collection, the flies were reared in a controlled climate chamber at a temperature of 27 ± 1 °C, relative humidity of 65 ± 5%, and a photoperiod of 14 L:10D. During rearing, adult Z. scutellata were fed an artificial diet consisting of a 1:1 mixture of beer yeast powder and sucrose. Once approximately 200 adult flies had been collected, individuals were frozen in liquid nitrogen and stored at −80 °C for subsequent analyses.
Genome survey and estimation
Genomic DNA (gDNA) was extracted from whole bodies of approximately ten adult female Z. scutellata individuals using a cetyltrimethylammonium bromide (CTAB) protocol. DNA integrity and purity were assessed via three complementary methods: (1) 1% agarose gel electrophoresis to evaluate fragment size distribution and check for RNA contamination; (2) spectrophotometric measurement with a NanoDrop™ instrument to determine A_260/A_280 and A_260/A_230 ratios; and (3) fluorometric quantification with a Qubit™ 2.0 system to obtain accurate concentration values. Samples that met DNA quality and quantity criteria, including sufficient concentration, high purity (A260/280 ≈ 1.8), and minimal degradation as assessed by agarose gel electrophoresis, were used for library preparation.
For genome survey sequencing, 1 µg of high-quality genomic DNA was fragmented to an average size of approximately 350 bp by ultrasonication using a Covaris system, in which controlled acoustic energy generates shear forces that randomly break DNA molecules, following the manufacturer’s recommended protocol. Libraries were constructed with the TruSeq Nano DNA HT Sample Preparation Kit (Illumina, USA) according to the manufacturer’s instructions. Briefly, fragmented DNA underwent end‐repair, 3′ adenylation, and ligation to indexed adapters, followed by PCR amplification and AMPure XP bead purification. Library size distribution was verified on an Agilent Bioanalyzer, and concentration was first estimated by Qubit™ before precise quantification via qPCR. Only libraries with an effective concentration >2 nM and an insert size centered at ~350 bp were sequenced. Paired‐end (2 × 150 bp) runs were performed on an Illumina NovaSeq 6000 platform (SmartGenomics Technology Institute, Tianjin, China), yielding 42.82 Gb of raw data (Table 1).
Raw reads were filtered with fastp15 (v0.23.2) to remove adapter sequences, reads containing >5 bp of adapter contamination, reads with ≥15% of bases below Q19, reads with >5% ambiguous bases (N), and orphaned mates. Quality‐filtered reads were then used for k–mer–based genome characterization. Jellyfish16 (v1.0.0) was employed to count 17-mers, and GCE17 (v1.0.2) was applied to estimate genome parameters. The 17-mer distribution exhibited a peak depth of 92, corresponding to a raw genome size of 406.84 Mb. After correction for sequencing errors and duplications, the estimated genome size was 404.37 Mb, with a heterozygosity rate of 2.31% and a repeat duplication rate of 38.70% (Table S1; Figure S1).
Pacbio HiFi sequencing and contig assembly
High–molecular-weight genomic DNA was extracted from approximately twenty adult female Z. scutellata individuals and further purified using AMPure PB beads (PacBio, Cat. No. 100-265-900) to eliminate contaminants. Library construction followed the SMRTbell Express Template Prep Kit 3.0 protocol (Pacific Biosciences). Briefly, genomic DNA was sheared to ~15 kb fragments using the Megaruptor system (Diagenode, Cat. No. B06010001), followed by size selection with AMPure PB beads, damage repair, and end polishing. SMRTbell adapters were then ligated to the processed fragments, which were further size-selected using a BluePippin system (Sage Science) to enrich the desired insert range. Library quality and concentration were validated using an Agilent 2100 Bioanalyzer (Agilent Technologies).
Sequencing was carried out on the PacBio Sequel II/IIe platform using SMRT Cells with a 30-hour run time, generating 19.72 Gb of raw long reads (Table 1). Raw polymerase reads were filtered using the SMRT Analysis suite to remove low-quality sequences (<50 bp, read quality <0.8, and self-ligated adapter artifacts). Circular Consensus Sequencing (CCS) reads were produced using SMRT Link v9.0 with parameters --min-passes=3 and --min-rq = 0.99, yielding high-fidelity (HiFi) reads.
Genome assembly was conducted using hifiasm18 (v0.16.1), which generated a set of primary contigs. These contigs served as the foundation for downstream scaffolding and polishing, ultimately resulting in a high-quality, chromosome-scale genome assembly.
Hi-C sequencing and chromosome-scale scaffold assembly
To generate chromosome‐scale scaffolds, we constructed a Hi‐C library from approximately twenty adult female individuals following a proximity‐ligation protocol. Intact nuclei were crosslinked with 1% formaldehyde, quenched with glycine, and then digested overnight with HindIII restriction enzyme. After end repair and biotin labeling of DNA termini, spatially proximate fragments were ligated using T4 DNA ligase to capture long‐range interactions. Crosslinks were reversed by proteinase K digestion, and the resulting DNA was purified. The purified DNA was sheared to 300–500 bp, and biotinylated junctions were enriched on Dynabeads® M‐280 Streptavidin (Life Technologies) prior to library construction. Paired‐end 150 bp sequencing on the Illumina NovaSeq 6000 platform yielded 55.8 Gb of raw Hi‐C data (Table 1).
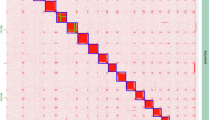
Hi‐C reads were trimmed and filtered as described for Illumina short‐read data and aligned to the preliminary contig assembly using HiCUP19 (v0.8.0). Uniquely mapping read pairs proximal to predicted HindIII sites were retained to generate a contact matrix. Contigs were polished twice with Pilon20 (v1.23) using the Illumina short reads, producing a draft assembly of 701.8 Mb in 685 contigs (N50 = 1.73 Mb; Table 1). We then applied ALLHiC21 (v0.9.8) to cluster, order, and orient contigs into linkage groups, using the Hi‐C contact map to resolve miss joins and confirm scaffold structure. Manual curation with Juicebox22 refined scaffold orientations and improved contact map consistency. The final assembly anchored 105 contigs (93.52% of the genome) into 6 chromosome‐scale scaffolds, achieving a scaffold N50 of 75.5 Mb (Fig. 1; Table 2 and S2).

Chromosome-scale Hi-C contact map and interspecific synteny of Z. scutellata. (a) Hi‐C interaction heatmap for the six assembled chromosomes of Z. scutellata. Strong intra‐chromosomal contacts appear as prominent diagonal blocks, whereas inter‐chromosomal interactions are comparatively weaker. Color intensity reflects normalized contact frequency. (b) Genome‐wide collinearity between Z. scutellata and Z. cucurbitae as determined by Satsuma. Syntenic blocks illustrate conserved genomic segments, highlighting structural conservation between the two Tephritid species.
Genome assembly quality assessment
Assembly completeness was assessed with BUSCO23 (v5.4.3) against the insecta_odb10 lineage dataset, which comprises 1,367 universal single‐copy orthologs. The analysis recovered 99.3% of BUSCO genes as complete, including 98.2% complete single‐copy and 1.1% complete duplicated orthologs, with only 0.5% fragmented and 0.2% missing (Table S3). These metrics demonstrate that our chromosome‐scale assembly is exceptionally complete and provides a robust foundation for downstream functional annotation, comparative genomics, and population‐level investigations.
Sexual chromosome identifies
To determine the sex chromosome of Z. scutellata, we performed a genome‐wide synteny analysis using Satsuma24 (v2.0), comparing our assembly to that of Z. cucurbitae (GenBank accession GCF_028554725.1; karyotype 5 + XY). Syntenic blocks were visualized with Circos25 (v0.69), which revealed extensive collinearity between chromosome 1 of Z. scutellata and the X chromosome of Z. cucurbitae (Fig. 2). This conserved syntenic relationship provides strong evidence that chromosome 1 in our assembly corresponds to the X chromosome in Z. scutellata.

Circos plot summarizing genome features of Z. scutellate. A genome-wide overview was generated using a 100 kb sliding window. Concentric tracks from the outermost (IV) to the innermost (I) circle denote: IV. Chromosome ideograms: each colored segment corresponds to one of the nine assembled chromosomes, with scale bars indicating genomic position in megabases. III. Protein-coding gene density: normalized counts of annotated genes per window, plotted as a histogram to highlight gene-rich and gene-poor regions. II. GC content density: proportion of guanine + cytosine bases calculated per window, displayed as a line plot to reveal regional GC variation. I. Repeat element density: percentage of bases masked as repetitive sequences in each window, shown as a heat-map track to illustrate the distribution of transposable elements and other repeats.
Genome annotation
High–depth transcriptomic data were generated from total RNA extracted from a single adult female Z. scutellata using the Qiagen RNeasy Plus Mini Kit. RNA integrity was confirmed on an Agilent 2100 Bioanalyzer (RIN > 7.0), and stranded mRNA libraries were constructed with the Illumina TruSeq Stranded mRNA Library Prep Kit. Paired-end (2 × 150 bp) sequencing on the NovaSeq 6000 platform yielded 7.38 Gb of clean transcriptome reads (Table 1), which provided empirical evidence for gene model refinement.
Repetitive elements were annotated with the HiTE26 pipeline, which integrates homology-based searches against known repeat libraries and de novo discovery algorithms. Approximately 34.38% of the Z. scutellata genome was classified as repetitive, comprising 3.24% long terminal repeats (LTRs), 5.88% long interspersed nuclear elements (LINEs), 0.07% short interspersed nuclear elements (SINEs), 15.50% DNA transposons, and 0.42% unclassified repeats (Table 3).
Protein-coding genes were predicted using eGAPX (v0.3.2, https://github.com/ncbi/egapx), combining ab initio algorithms with RNA-Seq alignments to accurately delineate exon–intron structures and untranslated regions. This produced 13,327 gene models encoding 14,117 transcripts. Predicted proteins were functionally annotated via BLASTP searches against NCBI NR and UniProt Swiss-Prot databases (E-value < 1 × 10−⁵), and assigned Gene Ontology (GO) terms and KEGG pathway identifiers to infer biological roles and metabolic functions (Table S4).
Non–coding RNAs were identified following repeat masking and removal of protein-coding loci. Transfer RNAs were detected with tRNAscan-SE27 (v2.0) under eukaryotic parameters. Ribosomal RNA genes were annotated by BLASTN alignment to curated invertebrate rRNA references (E-value < 1 × 10−5). Small nuclear and nucleolar RNAs were predicted using INFERNAL28 (v1.1.4) against Rfam29 14.9 profiles. In total, we annotated 432 tRNAs, 2,466rRNAs, 83 snoRNAs, and 65 miRNAs (Table 4). Together, these comprehensive annotations establish a robust genomic framework for functional and comparative studies in Z. scutellata.
Data Records
The genome sequencing and annotation data generated in this study have been deposited in multiple public repositories. At the National Genomic Data Center (NGDC), the genome assembly is available under the accession number of GWHGEAZ00000000.130 in the Genome Warehouse database31,32 (GWH, https://ngdc.cncb.ac.cn/gwh), with raw sequencing reads deposited in the Genome Sequence Archive33 (GSA, https://ngdc.cncb.ac.cn/gsa) under BioProject PRJCA041109. Specifically, Pacbio Hifi (CRR188411134), Illumina (CRR188411035), Hi-C (CRR188411236) and RNA-seq (CRR188411337) datasets are accessible through the GSA. In addition, all sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject PRJNA122541838. The Illumina, PacBio, and Hi-C datasets are available under accession numbers SRR3522705439, SRR3522705540, and SRR3522705641, respectively, while the RNA-seq data are deposited under accession number SRR3522705342. The chromosome-scale genome assembly is available in GenBank under accession JBLXXD000000000, with the corresponding assembly accession GCA_051201265.143. Furthermore, the assembly genome and annotated gene sets have also been deposited in the FigShare repository44 to facilitate open access and reuse by the research community.
Technical Validation
Sequencing data quality
The quality of raw Illumina paired-end reads was assessed using FastQC, confirming high base-calling accuracy and the absence of technical artifacts. Over 93% of bases achieved a Phred quality score of Q30 or above, and the GC content was uniformly distributed (~38.4%), consistent with characteristics of Tephritid genomes. After adapter trimming and quality filtering with fastp, a total of 41.44 Gb of high-quality short-read data was retained. PacBio HiFi sequencing produced long reads with a minimum of three polymerase passes (min-RQ ≥ 0.99), yielding a median base accuracy exceeding 99.5% (Table 1). This high-fidelity dataset provided a solid foundation for accurate and contiguous genome assembly.
Assembly consistency and completeness
K-mer analysis of the Illumina data using 17-mers revealed a distinct peak at depth 92. Genome size and heterozygosity were estimated using GCE, resulting in a predicted genome size of 404.37 Mb and a heterozygosity rate of 2.31%. These estimates closely matched the final Hi-C–scaffolded assembly size of 408.2 Mb, indicating minimal loss due to collapsed heterozygous regions or overrepresentation of repetitive sequences (Figure S1, Table S1). Hi-C contact matrices generated during scaffolding with ALLHiC revealed strong intra-chromosomal interaction patterns with minimal off-diagonal noise, confirming correct contig orientation and placement across six pseudochromosomes (Fig. 1). Additionally, synteny analysis demonstrated high chromosomal collinearity with Z. cucurbitae, further validating the structural integrity of the assembly (Fig. 2). BUSCO analysis using the insecta_odb10 dataset recovered 99.3% of expected conserved genes, including 98.2% single-copy and 1.1% duplicated BUSCOs, with only 0.7% missing or fragmented. This result indicates near-complete coverage of both coding and non-coding genomic regions (Table S3).
Annotation validation
Transcriptomic validation of gene models was performed by mapping RNA-Seq reads to the genome. The mapping achieved an overall alignment rate exceeding 95.4%, with more than 86% of reads spanning annotated exon–intron boundaries, confirming strong transcriptomic support. Annotation Edit Distance (AED) scores further evaluated the quality of gene models, with 92% of predicted loci exhibiting AED values ≤ 0.25, indicating high consistency between predicted and empirically supported gene structures. A total of 13,327 protein-coding genes were predicted in the Z. scutellatus genome, of which 91.58% (12,205 genes) were successfully annotated in the NCBI non-redundant (NR) database, indicating a high degree of sequence homology with known proteins. Swiss-Prot and Pfam annotations covered 53.67% and 82.91% of the genes, respectively, reflecting both curated functional matches and conserved protein domains. Additionally, 84.62% of genes were assigned to KOG categories, and 72.67% were associated with Gene Ontology (GO) terms, supporting the structural and functional integrity of the gene models. KEGG pathway annotation was assigned to 59.31% of the genes, enabling reconstruction of key metabolic and signaling pathways. These results collectively confirm the robustness of gene prediction and highlight the biological relevance of the Z. scutellatus gene repertoire (Table S4). Non-coding RNAs and repetitive elements were annotated using Rfam and Repbase, respectively, ensuring comprehensive identification of small RNAs and transposable elements (Table 4).
Data availability
All genome sequencing and annotation data generated in this study are publicly available. The chromosome-scale genome assembly is deposited in the Genome Warehouse (GWH) at the National Genomic Data Center under accession GWHGEAZ00000000.128, with raw PacBio HiFi, Illumina, Hi-C, and RNA-seq data available in the Genome Sequence Archive under BioProject PRJCA041109 and in the NCBI Sequence Read Archive under BioProject PRJNA122541836. The genome assembly is also available in GenBank under accession JBLXXD000000000 (assembly accession GCA_051201265.141), and the assembled genome and annotated gene sets are deposited in Figshare (https://doi.org/10.6084/m9.figshare.29322953.v1).
Code availability
Programs used in data processing were executed with default parameters unless otherwise specified in the Methods section. No custom code was employed for these analyses.
References
Gokulanathan, A., Mo, H. & Park, Y. Insights on reproduction‐related genes in the striped fruit fly, Zeugodacus scutellata (Hendel)(Diptera: Tephritidae). Archives of Insect Biochemistry and Physiology 115, e22064 (2024).
Liu, J.-H. et al. Complete mitochondrial genome of stripped fruit fly, Bactrocera (Zeugodacus) scutellata (Diptera: Tephritidae) from Anshun, Southwest China. Mitochondrial DNA Part B 2, 387–388 (2017).
Kim, Y. et al. Discrimination of different generations of Zeugodacus scutellata using age grading technique and their local genetic variation. Journal of Asia-Pacific Entomology 22, 908–915 (2019).
Kim, Y.-P. et al. Seasonal occurrence and damage of Bactrocera scutellata (Diptera: Tephritidae) in Jeonbuk province. Korean journal of applied entomology 49, 299–304 (2010).
Miyatake, T., Kuba, H. & Yukawa, J. Seasonal occurrence of Bactrocera scutellata (Diptera: Tephritidae), a cecidophage of stem galls produced by Lasioptera sp.(Diptera: Cecidomyiidae) on wild gourds (Cucurbitaceae). Annals of the Entomological Society of America 93, 1274–1279 (2000).
Al Baki, M. A., Vatanparast, M. & Kim, Y. Male-biased adult production of the striped fruit fly, Zeugodacus scutellata, by feeding dsRNA specific to Transformer-2. Insects 11, 211 (2020).
Han, H.-Y., Choi, D.-S. & Ro, K.-E. Taxonomy of Korean Bactrocera (Diptera: Tephritidae: Dacinae) with review of their biology. Journal of Asia-Pacific Entomology 20, 1321–1332 (2017).
Park, K. C., Jeong, S. A., Kwon, G. & Oh, H.-W. Olfactory attraction mediated by the maxillary palps in the striped fruit fly, Bactrocera scutellata: Electrophysiological and behavioral study. Archives of Insect Biochemistry and Physiology 99, e21510 (2018).
Al Baki, M. A. et al. Age grading and gene flow of overwintered Bactrocera scutellata populations. Journal of Asia-Pacific Entomology 20, 1402–1409 (2017).
Zhang, K.-J. et al. Comparison between different individuals of Bactrocera (Zeugodacus) Scutellata (Hendel)(Diptera: Tephtitidae) reveals differential mutation rates of mitochondrial genes. Mitochondrial DNA Part B 5, 1733–1734 (2020).
Jin, Y., Liu, X.-F. & Ye, H. Research overview on Bactrocera (Zeugodacus) scutellata (Hendel) (Diptera: Tephitidae). Biological Disaster Science 37, 191–197 (2014).
Li, X. Z. et al. Long-term monitoring of Bactrocera and Zeugodacus spp.(Diptera: Tephritidae) in China and evaluation of different control methods for Bactrocera dorsalis (Hendel). Crop Protection 182, 106708 (2024).
Jiang, F., Liang, L., Wang, J. & Zhu, S. Chromosome-level genome assembly of Bactrocera dorsalis reveals its adaptation and invasion mechanisms. Commun Biol 5, 25 (2022).
Wang, Y.-T. et al. Chromosome-level genome assembly of an agricultural pest Zeugodacus tau (Diptera: Tephritidae). Sci Data 10, 848 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marcais, G. & Kingsford, C. Jellyfish: A fast k-mer counter. Tutorialis e Manuais 1, 1038 (2012).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv:1308.2012 (2013).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18, 170–175 (2021).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Research 4 (2015).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963 (2014).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nature plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Gene prediction: methods and protocols 227–245 (2019).
Grabherr, M. G. et al. Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics 26, 1145–1151 (2010).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome research 19, 1639–1645 (2009).
Hu, K. et al. HiTE: a fast and accurate dynamic boundary adjustment approach for full-length transposable element detection and annotation. Nature Communications 15, 5573 (2024).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–964 (1997).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database. Nucleic acids research 31, 439–441 (2003).
NGDC Genome Warehouse (GWH). https://ngdc.cncb.ac.cn/gwh/Assembly/98044/show (2025).
CNCB NGDC Members & Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2025. Nucleic acids research 53, D30–D44 (2024).
Ma, Y. et al. The updated genome warehouse: enhancing data value, security, and usability to address data expansion. Genomics, Proteomics & Bioinformatics qzaf010 (2025).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics, Proteomics & Bioinformatics 19, 578–583 (2021).
NGDC Genome Sequence Archive (GSA). https://ngdc.cncb.ac.cn/gsa/browse/CRA026383/CRR1884111 (2025).
NGDC Genome Sequence Archive (GSA). https://ngdc.cncb.ac.cn/gsa/browse/CRA026383/CRR1884110 (2025).
NGDC Genome Sequence Archive (GSA). https://ngdc.cncb.ac.cn/gsa/browse/CRA026383/CRR1884112 (2025).
NGDC Genome Sequence Archive (GSA). https://ngdc.cncb.ac.cn/gsa/browse/CRA026383/CRR1884113 (2025).
NCBI Broproject. https://identifiers.org/ncbi/bioproject:PRJNA1225418 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR35227054 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR35227055 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR35227056 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR35227053 (2025).
NCBI Genome. https://identifiers.org/ncbi/insdc.gca:GCA_051201265.1 (2025).
Zhang, J. & Yin, C. A chromosome-scale genome assembly of the striped fruit fly Zeugodacus scutellatus (Diptera: Tephritidae). FigShare https://doi.org/10.6084/m9.figshare.29322953.v2 (2025).
Acknowledgements
This research was funded by the Key Investigation and Monitoring of Agricultural Alien Invasive Species project of the Ministry of Agriculture and Rural Affairs of China (No.13220138, No.13230120, No.019240117), the National Science Foundation of China (No. 32202315), and the Agricultural Alien Invasive Species Survey Project of Zhejiang Province (No.HT-CTZB-2022050535).
Author information
Authors and Affiliations
Contributions
J.M.Z. and C.L.Y. designed the project and coordinated the study, Y.X.W. and H.Y.Z. conducted the sampling and sequencing; C.L.Y. and S.X.Z. assembled the genome and annotated the genome; C.L.Y. and X.Y.J. performed the chromosomal syntheses analysis, comparative genomics analysis; M.L. performed the gene family identification; J.M.Z. and C.L.Y. drafted the manuscript, and Y.B.L. improved and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, JM., Jia, XY., Zhou, SX. et al. A chromosome-scale genome assembly of the striped fruit fly Zeugodacus scutellatus (Diptera: Tephritidae). Sci Data 13, 413 (2026). https://doi.org/10.1038/s41597-026-06828-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06828-w
