
Abstract
Utilizing non-human primates to study the role of human Tau and its related pathologies is logical and important due to their closer similarity to human brain structure and function. In our earlier research, we generated a transgenic cynomolgus monkey model expressing Tau (P301L) through lentiviral infection of monkey embryos. These monkeys exhibited age-dependent neurodegeneration and motor dysfunction. Single-nucleus RNA sequencing (snRNA-seq) is a powerful and promising technique for elucidating the cellular complexity and pathology across different tissues. However, single-cell data from non-human primate models of Tau pathology are currently nonexistent. In this study, we performed snRNA-seq on the hippocampus, striatum, and spinal cord of Tau (P301L) monkey, providing the first snRNA-seq atlas of multiple tissue regions in a non-human primate model that simulates human tauopathies. This will offer crucial data references for cross-species single-cell level studies of tau and its related pathologies.
Similar content being viewed by others

Background & Summary
Tauopathies are a category of neurodegenerative disorders, which are characterized by the dysfunction of tau and the consequent accumulation of its insoluble aggregates within the brain, as observed at autopsy1,2. Alzheimer’s disease (AD) and primary tauopathies (such as progressive supranuclear palsy [PSP] and frontotemporal dementia due to tauopathy [FTD]), are marked by progressive neurodegeneration3,4. The pathological burden of tau, in particular, has been shown to be closely linked to the severity of neurodegenerative pathologies and their associated clinical symptoms, thus rendering it a focal point in the development of new therapeutic interventions5,6,7.
To study tauopathies, particularly AD, researchers have developed various animal models to investigate their pathogenic mechanisms and explore potential therapeutic strategies. These models are crucial for understanding the complex biology of tau protein and its role in neurodegenerative diseases. The P301S Tau transgenic mouse model has been widely used to study tau pathology and its progression, offering insights into tau aggregation and neurofibrillary tangle formation8,9,10. Additionally, mouse models expressing a truncated tau form (Tau35) have emphasized the role of post-translational modifications in tau pathology1,11. While rodent models have been pivotal in advancing our mechanistic understanding of AD pathogenesis, their translational utility for clinical drug development has been largely disappointing12. A key limitation stems from profound interspecies divergences in genomic architecture, cellular metabolism, and neuroimmune responses, ultimately restricting their capacity to recapitulate the multidimensional complexity of human AD pathology13,14,15,16,17.
In view of the limitations inherent to mouse models, researchers have developed a series of non-human primate models that effectively mimic tau pathology observed in human AD patients. The vast majority of these models were created by stereotaxically injecting adeno-associated virus (AAV) into the brains of non-human primates to overexpress certain tau mutant forms, thereby inducing tau pathology13,18,19. In this context, our team has pioneered the development of a transgenic monkey model expressing Tau-P301L. This model effectively replicates tau pathology observed in AD patients and demonstrates progressive neurodegeneration20.
snRNA-seq is an advanced technology that provides deep insights into cellular transcript diversity and complexity. It enables the identification and characterization of various cell types within an organ or tissue, revealing their composition and functions. Current single-cell research in animal models of AD, especially those with tau pathology, mainly focuses on mice, with limited data from larger animals21,22.
In the present study, we employed 10x Genomics snRNA-seq to elucidate the cellular composition of multiple tissues, including the hippocampus, striatum, and spinal cord, in Tau-P301L transgenic monkey and wild-type control. The generation of snRNA-seq data encompassing 53,491 cells was successfully achieved. Following a rigorous process of quality control, a single-cell atlas was established. The atlas under consideration is composed of 36,623 cells, all of which are of a high quality.
While conventional research on AD has predominantly concentrated on the hippocampus, the spinal cord represents a critical yet historically underappreciated component of the AD pathological landscape. Compelling evidence from current literature indicates that the core pathological hallmarks of AD—specifically amyloid-beta deposition and Tau neurofibrillary tangles—are not confined to the brain but extensively involve the spinal cord23,24,25. Underscoring the significance of spinal cord investigation is essential; it not only elucidates the anatomical basis for the gait abnormalities, balance deficits, and motor dysfunction observed in patients prior to cognitive decline, but also advocates for redefining AD as a systemic disorder affecting the entire central nervous system26. Consequently, we have expanded our snRNA-seq dataset derived from the TauP301L macaque model to explicitly include spinal cord samples.
This represents the first snRNA-seq data from a non-human primate transgenic monkey model for AD. This data is crucial for future AD research, as it enables the investigation of similarities and differences in cellular pathological processes across AD animal models at various species levels. It provides new opportunities for deciphering cellular alterations during AD progression, particularly in the context of tau pathology.
Methods
Animal
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Yuanxi Biotech Inc., Guangzhou. The Tau-P301L transgenic monkeys, along with age- and gender-matched healthy control cynomolgus monkeys, were housed and behaviorally evaluated at Guangdong Landau Biotechnology Co. Ltd., an AAALAC-accredited facility. All animal-related protocols received prior approval from the IACUCs of both Guangdong Landau Biotechnology Co. Ltd. and Jinan University. This study was conducted in strict compliance with the “Guide for the Care and Use of Laboratory Animals (2011)” to ensure the welfare of the animals and the safety of personnel. The health and behavior of the monkeys were monitored daily by the husbandry staff and veterinarians.
Animals designated for tissue sampling were euthanized via intravenous administration of pentobarbital sodium, consistent with the AVMA Guidelines for the Euthanasia of Animals (2020). Vital signs were monitored until cessation, followed by immediate tissue harvest for snRNA-seq processing.
Tissue dissociation and preparation of single-cell suspensions
Approximately 500 mg of fresh-frozen tissue was dissected from the striatum, hippocampus, and spinal cord of transgenic monkey (Tau P301L) and wild-type control using sterile disposable surgical blades on dry ice (Table 1). Tissues were weighed and transferred to cold Dounce homogenizers containing ice-cold homogenization buffer (0.25 M sucrose, 5 mM CaCl₂, 3 mM MgAc₂, 10 mM Tris-HCl pH 8.0, 0.1 mM EDTA, 1 × protease inhibitors, 1 U/μL Ribolock RNase inhibitor). Homogenization was performed with 10 strokes using the loose (A) pestle followed by 10 strokes with the tight (B) pestle. The homogenate was filtered through a 70-μm cell strainer to remove debris.
Nuclei were purified by density gradient centrifugation. Filtered homogenate was mixed 1:1 with 50% iodixanol and layered over a 30–33% iodixanol cushion, then centrifuged at 10,000 × g for 20 min at 4 °C. After removing the myelin layer, nuclei were collected from the 30–33% iodixanol interface. The nuclear pellet was washed in nuclear resuspension buffer (phosphate buffer containing 0.04% BSA, 0.2 U/μL Ribolock RNase inhibitor, 500 mM mannitol, 0.1 mM PMSF) and centrifuged at 500 × g for 5 min at 4 °C.
Nuclei were quantified by trypan blue exclusion using a hemocytometer. Final suspensions were adjusted to 700–1200 nuclei/μL in resuspension buffer and immediately processed for 10x Genomics Chromium single-cell library preparation.
Chromium 10x genomics library and sequencing
The Illumina NovaSeq PE150 sequencing strategy was utilised to generate one lane of sequencing data, yielding approximately 120 gigabytes of raw data. The raw image files from high-throughput sequencing were processed using CASAVA for base calling, resulting in sequenced reads stored in FASTQ format. FASTQ is a prevalent text format for the storage of biological sequences alongside their corresponding quality scores. In data generated by the Illumina NovaSeq PE150 platform, both paired-end reads are 150 base pairs (bp) in length. For 10X Genomics libraries, the first 26 base pairs of read 1 contain cell barcode and UMI (unique molecular identifier) information, which is crucial for distinguishing the origin of read 2. It is considered that the bases following the initial 26 base pairs in read 1 are non-informative. In read 291 bp are regarded as informative data.
Barcode assignment, Unique Molecular Identifier (UMI) counting, and alignment
For initial data processing, we employed the standard 10x Genomics Cell Ranger pipeline with default settings for preliminary analyses. This process encompassed quality control of the FASTQ files, alignment to a customized medaka reference genome using STAR, demultiplexing of cellular barcodes, and UMI counting. Tables 2, 3 summarize the quality control metrics and output generated by Cell Ranger. The snRNA-seq data were mapped to the Ensembl genome of the crab-eating macaque (Macaca fascicularis, version 5.0).
Cell quality control
The output from Cell Ranger was imported into Scanpy27 for further dimensionality reduction, clustering, and analysis of the snRNA-seq data.
In the preliminary processing of snRNA-seq data, we implemented a series of stringent quality control measures to ensure high data quality and reliability. The specific filtering steps are as follows:
-
1.
Filtering Cells with Fewer than 200 Genes: Low-quality cells expressing fewer than 200 genes were removed.
-
2.
Filtering Potential Doublets: Cells expressing more than 6000 genes were excluded, as these are likely doublets (two cells captured together).
-
3.
Filtering Lowly Expressed Genes: Genes expressed in fewer than three cells were removed to minimize technical noise.
-
4.
Filtering Cells with Low Total Counts: Cells with fewer than 500 total counts were excluded to ensure sufficient transcript counts for analysis.
-
5.
Filtering High-count Outliers: Cells exhibiting total counts surpassing the 97th percentile were excluded from the analysis, as these cells could potentially represent doublets or possess anomalously elevated mRNA levels.
-
6.
Filtering Cells with High Mitochondrial Gene Proportions: Cells with mitochondrial gene proportions exceeding 15% were excluded, as these might be dead or dying cells.
-
7.
Filtering Cells with High Hemoglobin Gene Proportions: Cells with hemoglobin gene proportions exceeding 3% were filtered out to avoid interference from blood cells.
-
8.
Filtering Cells with High Ribosomal Gene Proportions: Cells with ribosomal gene proportions exceeding 10% were removed to reduce ribosomal RNA interference.
After completing all filtering steps, we obtained a high-quality snRNA-seq dataset. The final dataset consists of 36,623 cells and 20,683 genes, with an average mitochondrial gene proportion of 1.72% and an average gene count of 1,635 per cell.
Doublet detection
By implementing Scrublet28 with an expected doublet rate of 10% and taking batch effects into account, we effectively identified and removed doublets from our snRNA-seq data. This process significantly enhanced the quality of our dataset, ensuring more reliable and robust results in subsequent analyses. After removing the potential double cells, we obtained 35156 high quality cells.
Cell clustering
To identify distinct cell populations in snRNA-seq data, we employed scVI29, a deep generative model specifically designed for single-cell transcriptomics. By modeling latent variables for each batch, scVI addresses batch effects, ensuring that biological signals are retained while minimizing unnecessary technical variation. A k-nearest neighbors (k-NN) graph was constructed using the latent space, and the Leiden clustering algorithm was applied to this graph to identify distinct cell populations.
Differentially expressed genes analysis and marker genes analysis
We performed differential gene expression analysis across Leiden algorithm-identified clusters using the scanpy.tl.rank_genes_groups() function with default parameters. Statistical significance of expression differences was assessed using the Wilcoxon rank-sum test.
Cellular purity assessment
The purity and transcriptional homogeneity of identified cell clusters were evaluated using the ROGUE package30. ROGUE calculates an entropy-based metric to quantify the robustness of gene expression within clusters, where higher ROGUE scores (range: 0–1) indicate greater transcriptional homogeneity.
Public data sources for cross-species validation
The human and murine datasets utilized in this study were obtained from the GEO database. Specifically, the human sporadic early-onset Alzheimer’s disease (sEOAD) data and the P301L tauopathy mouse model data are accessible under accession numbers GSE272082 and GSE153895, respectively31,32.
Data Records
The snRNA-seq data generated in this study have been deposited in the NCBI BioProject database under accession number PRJNA132764. Raw sequencing data and detailed sample descriptions are available via the Sequence Read Archive (SRA) under accession SRP63104633, and BioSample accessions SAMN51289346–SAMN51289351. Additionally, the fully processed snRNA-seq datasets have been uploaded to the GEO and can be accessed under accession number GSE31717634. The processed gene lists, including differentially expressed genes, cell-type marker genes, and the top 200 signature genes for cross-species DAM-like microglial clusters, have been deposited in Zenodo (https://zenodo.org/records/18612605)35.
Technical Validation
Sequencing and quality assessment of the data
In this study, the 10x Genomics Chromium platform was employed to construct snRNA-seq libraries from the hippocampal, striatal, and spinal cord tissues obtained from both Tau-P301L transgenic monkey and WT control monkey. These libraries were subsequently sequenced using the Illumina NovaSeq PE150 sequencing system (Fig. 1). Analysis of the snRNA-seq data revealed that the number of reads obtained from the WT control monkey’s hippocampal, striatal, and spinal cord tissues were 475 million, 402 million, and 408 million, respectively. In contrast, the Tau-P301L transgenic monkey tissues yielded 484 million, 465 million, and 506 million reads for the hippocampus, striatum, and spinal cord, respectively. The detection rate of valid barcodes exceeded 94% across all tissues, and the proportion of reads successfully mapped to the genome surpassed 93%, underscoring the high quality of the data. Table 2 provides a detailed summary of the quality metrics for the sequencing process across all tissues. The estimated numbers of cells obtained from the hippocampus, striatum, and spinal cord tissues of WT monkey were 8,330, 4,923, and 6,323, respectively. In contrast, the corresponding estimates for the Tau-P301L transgenic monkey tissues were 6,801, 8,131, and 18,983. The median unique molecular identifier (UMI) count per cell in the WT monkey tissues was 1,636 for the hippocampus, 2,442 for the striatum, and 5,653 for the spinal cord. For the Tau-P301L transgenic monkey tissues, the median UMI counts per cell were 1,795, 1,842, and 3,875, respectively. Furthermore, the median number of genes detected per cell in the aforementioned tissues of WT monkeys was 806, 1,104, and 2,197, while in the Tau-P301L transgenic monkey tissues, it was 862, 871, and 1,949, respectively (Table 3). Collectively, these findings suggest that the sequencing process yielded high-quality data.

Single-nucleus transcriptomic profiling workflow. (a) Four-year-old TauP301L transgenic monkeys and age-matched wild-type (WT) controls were euthanized, and tissues (hippocampus, striatum, spinal cord) were collected. Nuclear isolation for snRNA-seq were prepared and subjected to snRNA-seq. Subsequent bioinformatic analyses included cell type identification.
Cell type identification
The cells procured as described underwent stringent quality control, with comprehensive parameters and procedures detailed in the “Cell quality control” and “Doublet detection” sections of the Methods. Following this, integration across various groups was performed using scVI. The classification of cells into 13 distinct clusters was achieved through the application of the Leiden algorithm, with visualization facilitated by UMAP (Fig. 2a).

Single-nucleus transcriptomic atlas and cellular composition analysis of the Tau-P301L monkey model. (a) The UMAP visualization of the cell clusters reveals a total of 13 distinct clusters following the clustering process, with the quantity of each cell type indicated on the right. The number of nuclei recovered for each cell type is indicated in the legend. (b) A stacked bar chart illustrating the proportion of various cell types in Tau transgenic monkey tissues in comparison to WT control tissues. (c) The dotplot shows marker genes in all clusters. Dot size represents the percentage of cells expressing the marker, and color intensity represents the average expression level. (d) Using the R package named ROGUE, check whether the given clustering is a pure cell group. High ROGUE scores indicate high purity and low heterogeneity within the identified cell clusters.
Annotation of the various clusters was performed using a selection of well-established marker genes, with the results corroborated by pertinent literature (Fig. 2c). Two distinct transcriptional states of microglia have been identified. In the microglia_1 state, there is a high expression of C1QA, C1QC, and CSF1R. Conversely, microglia_2 is characterized by elevated expression levels of GPNMB, ITGAX, and CHIT1. Notably, the number of GPNMB-positive microglia is increased in patients with AD. In transgenic AD models exhibiting significant neuronal loss, GPNMB levels rise in an age-dependent manner. Additionally, GPNMB has been shown to co-localize with various IBA1-positive microglial populations surrounding amyloid plaques. Currently, GPNMB is considered a marker of the activated state of microglia, a state observed exclusively under neurodegenerative conditions36,37. Through the analysis of bar charts depicting cell proportions, we observed an increase in the proportion of the microglia_2 cell population within the Tau transgenic monkey tissues (Fig. 2b). This finding suggests that microglia may exhibit distinct transcriptional characteristics in Tau transgenic monkeys. Astrocytes, choroid plexus epithelial cells, endothelial cells, fibroblasts, neurons, oligodendrocyte precursor cells (OPCs), oligodendrocytes, progenitor cells, Schwann cells, and T cells were identified utilizing well-established cellular markers. Furthermore, we consulted prior research to identify ventricular zone (VZ) cells through the expression of genes such as GFAP299, GFAP54, and TMEM23238. Figure 3a illustrates the top 20 marker genes identified for each distinct cell type, determined through the application of the Wilcoxon rank-sum test.

Identification of cell type-specific transcriptional signatures. (a) Top 20 marker genes identified for each distinct cell type.
Utilizing the ROGUE package in R, we assessed the even distribution and purity of the specified clusters. Our analysis revealed that the ROGUE values for each cell type cluster exceeded 0.75, indicating satisfactory cell quality and clustering purity (Fig. 2d).
Regional heterogeneity of glial responses to Tau pathology
Figures 4, 5 present the snRNA-seq differentially expressed genes (DEGs) in astrocytes, microglia, and oligodendrocytes from the hippocampus, striatum, and spinal cord of Tau-P301L monkeys, visualized via volcano plots and Venn diagrams. These figures highlight both cell type–specific transcriptional changes under Tau pathology and region-dependent transcriptional signatures within the same cell type.

Differential gene expression profiles in hippocampal and striatal glia of Tau-P301L monkeys. Volcano plots showing snRNA-seq differential expression (Tau-P301L vs WT). The x-axis is the change in the fraction of cells expressing each gene (Δpct = pct Tau-P301L – pct WT) and the y-axis is log2 fold change. Genes with FDR < 0.05 and |log2FC| > 0.25 are labeled; upregulated genes are pink and downregulated genes are blue. Panels: (a) hippocampal astrocytes; (b) hippocampal Microglia_1 subtype; (c) hippocampal oligodendrocytes; (d) striatal astrocytes; (e) striatal Microglia_1 subtype; (f) striatal oligodendrocytes.

Transcriptional landscape of spinal cord glia and cross-regional heterogeneity in the Tau transgenic monkey model. (a) DEGs in spinal cord astrocytes. (b) DEGs in spinal cord Microglia_1 subtype. (c) DEGs in spinal cord Microglia_2 subtype. (d) DEGs in spinal cord oligodendrocytes. (e–g) Venn diagrams illustrating the overlap of DEGs across the hippocampus, striatum, and spinal cord. Panels display the number and percentage of region-specific and shared DEGs in astrocytes (e), microglia_1 subtype (f), and oligodendrocytes (g).
Cross-species transcriptomic comparison validates the utility of the hippocampal P301L monkey model
To validate the biological relevance of this dataset and demonstrate its utility for translational research, we performed an integrated cross-species analysis focusing on the hippocampus (Table 4). We aligned our hippocampal monkey P301L data with high-quality public datasets from the hippocampus of human sporadic Early-Onset Alzheimer’s Disease (sEOAD) patients (GSE272082) and P301L transgenic mice (GSE153895)31,32. UMAP integration successfully resolved major cell lineages across all three species within the hippocampal formation (Fig. 6a,b). The identity of these clusters was further verified using canonical marker genes, which showed consistent expression patterns across species (Fig. 6c,d).

Cross-species transcriptomic comparison in hippocampus highlights a closer resemblance to human microglial pathology in the P301L monkey model compared to mice. (a,b) UMAP visualization of integrated single-nucleus RNA sequencing (snRNA-seq) datasets from (a) human sEOAD and controls, and (b) P301L transgenic mice and wild-type controls. Major cell types are annotated by color. (c,d) Dot plots validating cell type annotations using canonical marker genes for (c) the sEOAD human dataset and (d) the P301L mouse dataset. Dot size represents the fraction of expressing cells, and color intensity indicates mean expression levels. (e) A Venn diagram illustrating the overlap of DEGs in microglia across human sEOAD patients, P301L transgenic monkeys, and P301L mice. DEGs were filtered based on a statistical threshold of P < 0.05 and a log2 fold change magnitude (|log_2FC| > 0.25). (f) Rank-Rank Hypergeometric Overlap (RRHO) heatmaps displaying the concordance of transcriptional changes. Top panels: Comparison between Monkey P301L and Human sEOAD showing strong overlap in upregulated (bottom-left) patterns. Bottom panels: Comparison between Mouse P301L and Human sEOAD showing weaker concordance. (g) Dot plot comparing the expression patterns of specific disease-associated microglial genes. These genes are strongly upregulated in both human sEOAD and monkey P301L datasets but show discordant or limited responses in the mouse P301L model.
We utilized this integrated dataset to assess whether the monkey model captures the hippocampal transcriptional features of human pathology more accurately than the mouse model. A comparative analysis of DEGs in hippocampal microglia revealed that while there is a shared inflammatory response across species, the monkey P301L transcriptome exhibits a higher degree of overlap with human sEOAD. Rank-Rank Hypergeometric Overlap (RRHO) analysis further substantiated this, displaying a strong positive correlation in global gene expression patterns between the monkey and human hippocampal datasets (Fig. 6f, top panels), whereas the concordance between mouse and human was less pronounced (Fig. 6f, bottom panels). Notably, several key immune-associated genes (e.g., IL18, ST3GAL5, VSIG4) that were strongly upregulated in both human and monkey hippocampal samples showed discordant or limited responses in the mouse model (Fig. 6g). These findings underscore the high quality of the provided dataset and its potential to elucidate human-relevant pathogenic mechanisms specifically within the hippocampus.
Disease-associated microglia (DAM) signatures in the hippocampus
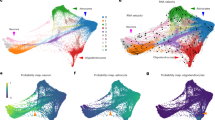
To further evaluate the dataset’s resolution in detecting subtle cell-state transitions within the hippocampus, we investigated the presence of DAM signatures. Using consensus Non-Negative Matrix Factorization (cNMF), we successfully identified distinct hippocampal microglial subpopulations in all three species (Fig. 7a–c). A specific DAM-like cluster was identified, which was characterized by the expression of established markers such as APOE, LPL, and SPP1 (Fig. 7d–f).

Cross-species comparison reveals conservation of human DAM signatures in the P301L monkey model. (a–c) UMAP visualization of microglial subpopulations identified by consensus Non-Negative Matrix Factorization (cNMF) clustering in P301L mice (a), human sEOAD patients (b), and P301L monkeys (c). (d–f) Dot plots characterizing the expression of key DAM marker genes (e.g., Apoe, Spp1) to identify DAM-like clusters in mice (d), humans (e), and monkeys (f). Dot size represents the fraction of expressing cells, and color intensity indicates average expression levels. (g–i) Stacked bar charts showing the proportional shifts of microglial clusters between control (WT/Control) and disease (P301L/sEOAD/Tau) conditions. Arrowheads indicate the expansion of DAM-like populations in the disease state across all three species. (j) UpSet plot quantifying the intersection of DEGs (Top200) in DAM-like clusters across mouse, human, and monkey. Note that the monkey model shares a larger number of unique DAM signature genes with humans (23 genes) compared to the mouse model (14 genes). (k) Detailed lists of the overlapping genes corresponding to the intersections shown in (j), highlighting the specific orthologous genes conserved between pairwise and three-way comparisons.
Consistent with pathological progression, the proportion of cells within this hippocampal DAM-like cluster was expanded in the disease groups (P301L/sEOAD) compared to controls across all species (Fig. 7g–i). However, the specific gene composition of this signature revealed significant species-specific differences. We quantified the intersection of the top 200 DEGs in the hippocampal DAM-like clusters (Fig. 7j) and found that the P301L monkey model shares a larger number of unique orthologous signature genes with humans (23 genes, including DDX5, CXCR4, and RPS family genes) compared to the mouse model (14 genes) (Fig. 7k). These validation steps confirm that the hippocampal dataset provided here possesses sufficient depth and biological fidelity to support advanced cross-species investigations.
Our analysis of the snRNA-seq data elucidated the heterogeneous cell populations present in the transgenic Tau-P301L monkey. While we acknowledge the constraints regarding biological replicates and the lower recovery of neuronal nuclei—factors attributable to the scarcity of transgenic primate models and the technical challenges of early isolation protocols—this dataset uniquely captures robust and evolutionarily conserved glial signatures that are frequently divergent in rodent models. This dataset thus establishes a foundational framework for identifying cell-type-specific responses within these critical tissues, offering a rare and valuable resource for translational inquiry. It holds significant potential for advancing the study of Tau pathology and facilitating high-fidelity cross-species investigations, thereby addressing the critical gap in current single-cell data which predominantly relies on murine models.
Data availability
The single-nucleus RNA sequencing (snRNA-seq) data generated in this study have been deposited in the NCBI BioProject database under accession number PRJNA132764. Raw sequencing data and detailed sample descriptions are available via the SRA under accession SRP63104633, and BioSample accessions SAMN51289346–SAMN51289351. Additionally, the fully processed snRNA-seq datasets have been uploaded to the GEO and can be accessed under accession number GSE31717634. The processed gene lists, including differentially expressed genes, cell-type marker genes, and the top 200 signature genes for cross-species DAM-like microglial clusters, have been deposited in Zenodo (https://zenodo.org/records/18612605)35.
Code availability
It is important to note that no specific custom code was employed during the analysis of the current dataset. The analyses and image generation were conducted using the following open-access software programs: Cell Ranger (3.0.1), Scanpy (1.11.3)27, Omicverse (1.7.5)27, ROGUE (1.0)30, and Seurat (5.4.0)39. All analyses were implemented in R (4.4.3) and Python (3.10.13).
References
Zhang, Y., Wu, K.-M., Yang, L., Dong, Q. & Yu, J.-T. Tauopathies: New perspectives and challenges. Mol. Neurodegener. 17, 28 (2022).
Samudra, N., Lane-Donovan, C., VandeVrede, L. & Boxer, A. L. Tau pathology in neurodegenerative disease: Disease mechanisms and therapeutic avenues. J. Clin. Invest. 135 (2023).
Langerscheidt, F. et al. Genetic forms of tauopathies: Inherited causes and implications of alzheimer’s disease-like TAU pathology in primary and secondary tauopathies. J. Neurol. 271, 2992–3018 (2024).
Hallmarks of neurodegenerative diseases. Cell 186, 693–714 (2023).
Samudra, N., Lane-Donovan, C., VandeVrede, L. & Boxer, A. L. Tau pathology in neurodegenerative disease: Disease mechanisms and therapeutic avenues. J. Clin. Invest. 133 (2023).
Targeting tau. Clinical trials and novel therapeutic approaches. Neurosci. Lett. 731, 134919 (2020).
Creekmore, B. C., Watanabe, R. & Lee, E. B. Neurodegenerative disease tauopathies. Annu. Rev. Pathol. 19, 345–370 (2024).
Yoshiyama, Y. et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007).
Schweighauser, M. et al. Cryo-EM structures of tau filaments from the brains of mice transgenic for human mutant P301S tau. Acta Neuropathol. Commun. 11, 160 (2023).
Wenger, K. et al. Common mouse models of tauopathy reflect early but not late human disease. Mol. Neurodegener. 18, 10 (2023).
Guo, T., Dakkak, D., Rodriguez-Martin, T., Noble, W. & Hanger, D. P. A pathogenic tau fragment compromises microtubules, disrupts insulin signaling and induces the unfolded protein response. Acta Neuropathol. Commun. 7, 2 (2019).
Polis, B. & Samson, A. O. Addressing the discrepancies between animal models and human alzheimer’s disease pathology: Implications for translational research. J. Alzheimers Dis. 98, 1199–1218 (2024).
He, F. et al. A mass-producible macaque model displays a durable alzheimer-like cognitive deficit and hallmark amyloid-β/tau/neurofilament light chain pathologies. J. Alzheimer’s Dis. 105, 1432–1446 (2025).
Esquerda-Canals, G., Montoliu-Gaya, L., Güell-Bosch, J. & Villegas, S. Mouse models of alzheimer’s disease. J. Alzheimers Dis. JAD 57, 1171–1183 (2017).
Chen, Z.-Y. & Zhang, Y. Animal models of alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 43, 1026–1040 (2022).
Zakaria, R. et al. Lipopolysaccharide-induced memory impairment in rats: A model of alzheimer’s disease. Physiol. Res. 66, 553–565 (2017).
Puzzo, D., Gulisano, W., Palmeri, A. & Arancio, O. Rodent models for alzheimer’s disease drug discovery. Expert Opin. Drug Discov. 10, 703–711 (2015).
Jiang, Z. et al. A nonhuman primate model with alzheimer’s disease-like pathology induced by hippocampal overexpression of human tau. Alzheimers Res. Ther. 16, 22 (2024).
Beckman, D. et al. A novel tau-based rhesus monkey model of alzheimer’s pathogenesis. Alzheimers Dement. J. Alzheimers Assoc. 17, 933–945 (2021).
Tu, Z. et al. Tauopathy promotes spinal cord-dependent production of toxic amyloid-beta in transgenic monkeys. Signal Transduct. Target. Ther. 8, 358 (2023).
Ali, M. et al. Temporal transcriptomic changes in the THY-Tau22 mouse model of tauopathy display cell type- and sex-specific differences. Acta Neuropathol. Commun. 13, 93 (2025).
Kim, D. W. et al. Amyloid-beta and tau pathologies act synergistically to induce novel disease stage-specific microglia subtypes. Mol. Neurodegener. 17, 83 (2022).
Dugger, B. N. et al. The distribution of phosphorylated tau in spinal cords of alzheimer’s disease and non-demented individuals. J. Alzheimers Dis. JAD 34, 529–536 (2013).
Lorenzi, R. M. et al. Unsuspected involvement of spinal cord in alzheimer disease. Front. Cell. Neurosci. 14 (2020).
Xie, Q., Zhao, W.-J., Ou, G.-Y. & Xue, W.-K. An overview of experimental and clinical spinal cord findings in alzheimer’s disease. Brain Sci. 9, 168 (2019).
Zhou, Y. et al. Spinal cord tau pathology induces tactile deficits and cognitive impairment in alzheimer’s disease via dysregulation of CCK neurons. Nat. Neurosci. 1–16, https://doi.org/10.1038/s41593-025-02137-4 (2025).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Liu, B. et al. An entropy-based metric for assessing the purity of single cell populations. Nat. Commun. 11, 3155 (2020).
Zhao, Z. GEO. https://identifiers.org/geo/GSE272082 (2025).
Lee, S., Meilandt, W. J., Friedman, B. A., Bohlen, C. J. & Hansen, D. V. GEO. https://identifiers.org/geo/GSE153895 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP631046 (2026).
Tu, Z. et al. GEO. https://identifiers.org/geo/GSE317176 (2026).
Han, B., Ouyang, W., Chen, Y., Chen, D., & Liang, W. Single-nucleus RNA sequencing dataset of diverse tissues from wild-type monkey and Tau-P301L transgenic monkey, Zenodo, https://doi.org/10.5281/zenodo.18612605 (2026).
Martins-Ferreira, R. et al. The human microglia atlas (HuMicA) unravels changes in disease-associated microglia subsets across neurodegenerative conditions. Nat. Commun. 16, 739 (2025).
Hüttenrauch, M. et al. Glycoprotein NMB: A novel alzheimer’s disease associated marker expressed in a subset of activated microglia. Acta Neuropathol. Commun. 6, 108 (2018).
Andersen, J. et al. Single-cell transcriptomic landscape of the developing human spinal cord. Nat. Neurosci. 26, 902–914 (2023).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Acknowledgements
The author thanks Xiaojiang Li and Shihua Li from Jinan University. This work was supported by the National Key Research and Development Program of China (2021YFA0805300,2021YFA0805200), the National Natural Science Foundation of China (32170981,82171244, 81922026).
Author information
Authors and Affiliations
Contributions
Z.T., S.Y. designed the research. All authors performed the research. B.H. wrote the paper, and Z.T., S.Y. edited the paper. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, B., Chen, Y., Ouyang, W. et al. Single-nucleus RNA sequencing dataset of diverse tissues from wild-type monkey and Tau-P301L transgenic monkey. Sci Data 13, 497 (2026). https://doi.org/10.1038/s41597-026-06882-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06882-4
