
Abstract
Gynostemma guangxiense X. X. Chen & D. H. Qin, belonging to the family Cucurbitaceae, is a perennial creeping herbaceous plant endemic to China with potential medicinal and health value. Here, we report the high-quality chromosome-level genome of G. guangxiense obtained by integrating Illumina short read, PacBio high-fidelity (HiFi) long read, Hi-C, and RNA-Seq technologies. The genome is anchored to 11 pseudochromosomes with a total size of 565.18 Mb, with a scaffold N50 of 52.63 Mb, achieving a complete BUSCO of 98.00%. Furthermore, we identified 27,527 protein-coding genes, of which 97.75% were functionally annotated. This genome provides an important molecular foundation for adaptive evolution, genetic conservation, and effective development of valuable medicinal plant resources within the Gynostemma genus.
Similar content being viewed by others

Background & Summary
Gynostemma guangxiense X. X. Chen & D. H. Qin (Cucurbitaceae) is a climbing leathery vine endemic to the limestone mountain forests in Guangxi Province, China. Its pedate leaves comprise (3-)5-7 ovate-elliptic or obovate leaflets that are nearly glabrous. Although similar in growth habit and leaf morphology to G. pentaphyllum, it differs in its female flowers, which form 1-2-flowered cymes, and in its trigonous-spherical fruits1 (Fig. 1). Unlike G. pentaphyllum, G. guangxiense has a sweet taste, and its whole herb is traditionally used to treat hepatitis and chronic bronchitis2. Gynostemma is the only known genus besides Panax that produces dammarane-type saponins3. Gypenosides II, IV, VIII, and XII are structural homologs of ginsenosides Rb1, Rb2, Rd1, and Rf2, respectively4.

Morphology and genomic features of G. guangxiense. (a) habitat; (b) leaves; (c) male flowers (panicles); (d) female flowers (cymose); (e) A scan of the herbarium specimen of G. guangxiense (Collector: Xiao Zhang and Yuemei Zhao; Collection site: Longzhou, Chongzuo, Guangxi, China; Environmental conditions: Under the forests on the hillside).
As an important sister species of G. guangxiense, G. pentaphyllum is a traditional Chinese herb having potential in treating hypertension, hyperlipidemia, and inflammation5,6, with additional health benefits including anticancer and immunomodulatory effects7,8. These medicinal value generate significant interest in the plants of the Gynostemma genus. However, the exclusive reliance on the widely distributed G. pentaphyllum has caused overexploitation and depletion of wild resources, leading to its classification as a national second-class protected species9. Comprehensive physiological, biochemical, and molecular studies across Gynostemma species are crucial for enhancing conservation efforts and broadening therapeutic applications. These researches will also resolve taxonomic uncertainties within this genus.
Previous metabolic studies on G. guangxiense have identified high leaf saponin and flavonoid content10,11, but reports on polysaccharides are scarce. Among the essential trace elements in the human body, Fe, Mn, Zn and Cu are the most important. These four elements are more abundant in G. guangxiense than in G. pentaphyllum, which has great development and utilization value12,13. Current genomic studies on G. guangxiense primarily cover chloroplast genome characterization14 and ISSR-PCR-based phylogenetics15. With the development of sequencing technologies, an increasing number of medicinal plant genomes have been sequenced and assembled16. These studies have laid a good molecular foundation for the identification and utilization, quality screening, biosynthesis of important components, and genetic improvements of medicinal plant species.
Here, we present a high-quality chromosome-level genome assembly of G. guangxiense obtained by integrating Illumina short read, PacBio high-fidelity (HiFi) long read, Hi-C, and RNA-Seq technologies. The assembled genome size was 565.18 Mb with a scaffold N50 of 52.63 Mb, achieving a complete BUSCO score of 98.00%. A total of 562.11 Mb (99.45%) of the assembled sequences was anchored to 11 pseudochromosomes. Genome annotation predicted 27,527 protein-coding genes, and the ratio of TE repetitive sequences was 67.38%. In conclusion, this reference genome of G. guangxiense provides valuable information that lays an important molecular foundation for adaptive evolution, genetic conservation, and the effective development of important medicinal plant resources within the Gynostemma genus in the future.
Methods
Sample collection and sequencing
Healthy plant material was obtained from a female individual of G. guangxiense growing in Chongzuo, Guangxi, China (22.35°N, 106.86°E), and a voucher herbarium specimen was identified and deposited at the Institute of Botany of Shaanxi Province under the voucher number LB2006921 (Fig. 1e). Genomic DNA was extracted from fresh young leaves following the protocol of the DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany), and randomly fragmented. A paired-end short read (150 bp) library was constructed and sequenced on the Illumina NovaSeq platform under 250 bp - 700 bp insert. Approximately, 33.64 Gb (59.43 × coverage) of short read data were obtained. For HiFi sequencing (15 - 20 kb insert size), a 20 kb library was constructed following the protocol for the PacBio Revio platform, and circular consensus sequencing (CCS) was performed (Table 1). A total of 24.58 Gb of HiFi long clean reads with an N50 of 19,981 bp were obtained for de novo assembly (Table 1). A Hi-C library was also sequenced on an Illumina NovaSeq platform with paired-end reads of 150 bp.The experiment processes were including (1) live samples were treated with 1–3% formaldehyde at room temperature for 10–30 minutes; (2) cutting the genome with Thermo Scientific Hindlll restriction enzyme, and then repair the end and add biotin; (3) the interacting fragments were linked using T4 DNA ligase to form a ring; (4) using ultrasound to break the fragment again and the biotin-containing fragments were captured using magnetic beads to create libraries and sequenced. Finally, 62.32 Gb reads (110.10 × coverage) (Table 1) were generated. Total RNA was extracted from five tissues (terminal buds, mature leaves, stems, flowers and fruits) using TRNzol Universal Reagent (TIANGEN BIOTECH DP424), and uniformly mixed after leveling the concentration. The concentration of the extracted nucleic acids was determined with NanoDrop spectrophotometer (Qubit 4.0) and integrity was detected with Qsep 400. The RNA-seq library was constructed and sequenced on the Illumina NovaSeq platform with 150 bp paired-end reads, generating a total of 10.68 Gb of clean data for genome annotation (Table 1).
Genome size estimation
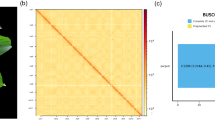
To determine the chromosomal ploidy of the sample, flow cytometry analysis was first conducted using a Partec CyFlow Space system (Jindi Future Biotechnology, Beijing, China), and a smudge plot was employed. Illumina short reads were filtered using fastp v0.23.417 software to remove adapters and low-quality reads. Thereafter, a total of 30.29 GB clean reads were used for genome survey analysis. A 31-bp k-mer with quality-filtered reads was counted and satisfied using KMC v3.2.118 to calculate genome size, repeat content, and heterozygosity. Subsequently, the K-mer data were fitted and analyzed using the skewed normal distribution model in GenomeScope v2.019 software. The final genome evaluation results showed that the genome size of G. guangxiense was approximately 554 Mb, with a heterozygosity of 4.21% and a repeat content of 53.5%, indicating that it was a complex genome (Fig. 2a).

Genome size estimation and Hi-C heatmap of G. guangxiense. (a) Genome size estimation by 17-K-mer analysis. (b) Heatmap of pseudochromosomes after Hi-C assisted assembly.
Genome assembly
After quality control, the PacBio HiFi reads were assembled into contigs using the Hifiasm v0.19.820 software with default parameters to obtain a preliminary assembly version of the genome, which was 803.27 Mb in size with a contig N50 of 44.97 Mb. The Hi-C clean reads were then aligned to the reference genome using Juicer v1.6 software21 with default parameters. To obtain valid pair reads, noisy reads, including low-quality reads, duplicated reads, single-ended reads, and reads of three or more positions aligned on the reference genome, were filtered, automatically clustered, sorted, and oriented using the YaHS software22 to generate Hi-C and assembly files. We then used Juicebox v1.11.0823 to manually inspect and adjust the draft assembly, and visualized the Hi-C contact maps of the genome assembly. Finally, approximately 562.11 Mb of scaffold was anchored to 11 longest scaffolds that were identified as pseudochromosomes, featuring a contig and scaffold N50 value of 47.45 Mb and 52.63 Mb, respectively (Tables 2, 3, Fig. 2b). The complete BUSCO score for the assembly was 98.00% (Table 4). Although this complete genomic sequences was high-quality, there were still some gaps, such as at the end of Chr1 and in the middle of Chr2 and Chr4 (Table 3, Fig. 2b). These regions have high GC content and were repeat-rich regions which may represent potential centromeric and telomeric regions (Fig. 3).

Genome map of G. guangxiense. (a) The 11 pseudochromosomes. (b) GC content. (c) Total repeat sequences density. (d) TE density. (e) TR density. (f) Gene density. (g) Intragenomic collinearity.
Annotation of repeat sequences
Repeat sequences constitute a substantial portion of the G. guangxiense genome (70.59%) and can be broadly classified into tandem repeats (TRs) and transposable elements (TEs) according to their distribution in the genome. First, Tandem Repeats Finder v. 4.0924 was used to search for TRs, and LTR elements were investigated using LTR FINDER v1.0725. And then, a non-redundant TE library of the G. guangxiense genome was constructed by extensive de novo TE annotator (EDTA) v1.026. TE repeat sequences were predicted using RepeatMasker27 with the default parameters. Finally, the results showed that the predicted proportion of TE repeat sequences was 67.38%, with long terminal repeats (LTRs) and long interspersed nuclear elements (LINEs) accounting for 44.55% and 2.05% of the genome, respectively (Table 5).
Annotation of non-coding RNAs
Based on the assembled genome sequence, software INFERNAL v1.1.428 trained on Rfam v14.829 wes used to predict the non coding RNA (ncRNA) of the genome. Meanwhile, tRNAscan-SE v2.0030 with parameter “--thread 4 -E -I” was used to predict tRNA, and RNAmmer v1.2031 with parameter “-S euk -m lsu,ssu,tsu -gff” was used to build models to predict rRNA and its various subunits. The final results were further integrated to obtain the prediction results of ncRNA in the G. guangxiense genome. In total, 696 tRNA, 4,282 rRNA and 692 ncRNA were indentified, respectively (Table 6).
Gene prediction and functional annotation
The presence of repeat sequences often makes it difficult to predict and annotate encoding genes. In this study, we shielded the TE sequences before predicting the encoding genes. Protein-coding gene annotation was performed using a combination of ab initio-, homology-, and transcriptome-based prediction methods. For ab initio prediction, Augustus v3.5.032 and GeneMark-ES v5.133 were employed to predict protein-coding genes. Second, the protein files of five species (G. pentaphyllum, Momordica charantia, Cucumis sativus, Nicotiana tabacum and Arabidopsis thaliana) were downloaded from the National Center for Biotechnology Information (NCBI) and aligned to the query genome as homologous proteins using GeMoMa v1.9034. Third, quality-controlled RNA-Seq data from the five tissues were mapped onto assembled genomes using HISAT2 v.2.2.135. Based on the alignment results, the mapped reads were assembled using StringTie v1.3.3b36, and PASA v2.5.237 was used to predict the UTR and variable splicing regions of the initially obtained gene set. To obtain the final non-redundant gene set, Evidence Modeler v1.1.138 was used to combine the three gene datasets with weights of 1, 1, 5, and 10 for GeneMark, Augustus, GeMoMa, and PASA, respectively. We also assessed the completeness of the gene set based on comparisons with the plant single-copy ortholog gene database (Embryophyta_odb10) using BUSCO v4.0.639. The results showed that 96.59% of the Embryophyta odb10 gene set was completely covered by genome annotations (Table 4). The gene sets obtained from gene structure annotations were searched against six known protein databases including the NCBI non-redundant protein (NR), Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), Eukaryotic Orthologous Groups (KOG), InterPro, and Swiss-Prot using BLAST v2.2.3140 with an e-value less than 1e-5. In total, 27,527 protein-coding genes were predicted, of which 97.75% were functionally annotated (Tables 2, 7).
Data Records
The clean Illumina reads, PacBio HiFi reads, Hi-C reads and RNA-seq reads for the G. guangxiense genome has been deposited in the NCBI Sequence Read Archive (SRA) database under accession number SRR3203022941, SRR3203022842, SRR3203022743, and SRR3203022644, respectively, under BioProject accession number PRJNA120968645. The final chromosome assembly is available in the National Center for Biotechnology Information (NCBI) GenBank under the accession No. JBLAST00000000046. The genome and its repeat annotation for G. guangxiense have been uploaded to figshare under accession number 2828317447 and 3008636248. Details and summary of functional annotation of G. guangxiense has been uploaded to figshare under accession number 3020430149.
Technical Validation
The completeness of the assembly and annotation was assessed based on the Embryophyta odb10 database using BUSCO v4.0.6, with default parameters. The assembly completeness of BUSCO was 98.00% (n = 1,614), and the annotated proteins completeness of BUSCO was 96.59% (n = 1,614) (Table 4). Clean Illumina, HiFi and RNA-Seq reads were mapped onto the assembled genome using BWA v0.7.1550, minimap2 v2.2851, and HISAT235 to assess the genomic integrity and accuracy. The mapping reads were 99.68%, 99.94%, and 99.39%, respectively. These results indicated that the G. guangxiense genome assembly was of high quality.
Data availability
The final chromosome assembly of G. guangxiense is available in the GenBank under the accession No. JBLAST000000000. The clean sequencing reads generated from three platform-specific sequencing runs, along with the final genome assembly, have been deposited in the NCBI Sequence Read Archive (SRA, accession numbers: SRR32030226 - SRR32030229).
Code availability
The software utilized in this study were executed in strict adherence to the official guidelines of published bioinformatics programs. Anything not mentioned in Methods was run with default settings. No custom codes were used.
References
Chen S., Lu A., & Charles J. Flora of China. Vol. 19. Beijing: Missouri Botanical Garden Press. (2011).
Chen, X. X. & Qin, D. H. A new species of the genus Gynostemma from Guangxi. Acta Botanica Yunnanica. 10(4), 495–496 (1988).
Chen, Q. et al. Transcriptome sequencing of Gynostemma pentaphyllum to identify genes and enzymes involved in triterpenoid biosynthesis. International Journal of Genomics. 2016, 1–10 (2016).
Kao, T., Huang, S., Inbaraj, B. & Chen, B. Determination of flavonoids and saponins in Gynostemma pentaphyllum (Thunb.) Makino by liquid chromatography-mass spectrometry. Analytica Chimica Acta. 626, 200–211 (2008).
Gou, S. H. et al. Anti-atherosclerotic effect of Fermentum Rubrum and Gynostemma pentaphyllum mixture in high-fat emulsion-and vitamin D3-induced atherosclerotic rats. Journal of the Chinese Medical Association. 81, 398–408 (2018).
Babich, O. et al. Medicinal plants to strengthen immunity during a pandemic. Pharmaceuticals. 13, 313–314 (2020).
Li, Y. et al. Anti-cancer effects of Gynostemma pentaphyllum (thunb.) makino (jiaogulan). Chinese Medicine. 11, 43–45 (2016).
Choi, K. T. Botanical characteristics, pharmacological effects and medicinal components of korean Panax ginseng C. a Meyer. Acta Pharmacologica Sinica. 29(11), 09–18 (2008).
Li, Z. H. et al. A review on studies of systematic evolution of Gynostemma Bl. Acta Botanica Boreali-Occiden-talia Sinica. 32, 2133–2138 (2012).
Liu, S., Lin, R. & Hu, Z. Comparison of stem and leaf structures and total gypenosides among 5 species of Gynostemma. Journal of Fujian Agriculture and Forestry University (Natural Science Edition). 35(5), 495–499 (2006).
Jiang, W., Zhou, Y. & Li, J. Assaying of total flavonoids in 6 kinds of Gynostemma made in Guangxi. Chinese Pharmacy. 7(1), 74–75 (2006).
Li, X., Liu, S., Yi, L. & Li, C. Seasonal variations in the contents of total saponins, total flavonoids and mineral elements of three species of the genus Gynostemma. Journal of Chinese Medicinal Materials. 35(1), 26–30 (2012).
Peng, X., Wang, T., Luo, Z. & Liu, S. Nutritional components of three species of Gynostemma (Cucurbitales: Cucurbitaceae). Journal of Mountain Agriculture and Biology. 36(1), 89–91 (2017).
Zhao, Y., Zhang, X., Zhou, T., Chen, X. & Ding, B. Complete chloroplast genome sequence of Gynostemma guangxiense: genome structure, codon usage bias, and phylogenetic relationships in Gynostemma (Cucurbitaceae). Brazilian Journal of Botany. 46, 351–365 (2023).
Wang, C. et al. Identification of seven plants of Gynostemma BL. by ISSR-pcr. Chinese Traditional and Herbal Drugs. 39(4), 588–591 (2008).
Zhang, X. et al. Diploid chromosome-level reference genome and population genomic analyses provide insights into Gypenoside biosynthesis and demographic evolution of Gynostemma pentaphyllum (Cucurbitaceae). Horticulture Research. 10(1), uhac231 (2023).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884–i890 (2018).
Deorowicz, S., Kokot, M., Grabowski, S. & Debudaj-Grabysz, A. KMC 2: fast and resource-frugal k-mer counting. Bioinformatics. 31(10), 15 (2015).
Vurture, G. W. et al. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics. 33(14), 2202–2204 (2017).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods. 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for Analyzing loop-resolution hi-C experiments. Cell Systems. 3, 95–98 (2016).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics. 39, btac808 (2023).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems. 3, 99–101 (2016).
Zhao, X. & Hao, W. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research. 35, 265–268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research. 27, 573–580 (1999).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome biology. 20, 275–259 (2019).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research. 110, 462–467 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29(22), 2933–5 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Research. 33(Database issue), D121–4 (2005).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research. 25(5), 955–64 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research. 35(9), 3100–8 (2007).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research. 34, 435–439 (2006).
Alexandre, L., Burns, P. D. & Mark, B. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Research. 42, e119 (2014).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods in Molecular Biology. 1962, 161–177 (2019).
Kim, D., Langmead, B. & Salzberg, S. HISAT: a fast spliced aligner with low memory requirements. Nature Methods. 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA - seq reads. Nature Biotechnology. 33, 290–295 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology. 9, R7 (2008).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Altschul, S. et al. Basic local alignment search tool. Journal of Molecular Biology. 215, 403–410 (1990).
NCBI Sequence Read Archive,https://identifiers.org/ncbi/insdc.sra:SRR32030229 (2025).
NCBI Sequence Read Archive,https://identifiers.org/ncbi/insdc.sra:SRR32030228 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32030227 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32030226 (2025).
NCBI National Genomics Data Center (NGDC) database https://identifiers.org/ncbi/bioproject:PRJNA1209686 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBLAST000000000 (2025).
Zhang, X. The genome annotation of Gynostemma guangxiense. figshare. https://doi.org/10.6084/m9.figshare.28283174.v1 (2025).
Zhang, X. The repeat annotation of Gynostemma guangxiense. figshare. https://doi.org/10.6084/m9.figshare.30086362.v1 (2025).
Zhang, X. Details and summary of functional annotation of Gynostemma guangxiense. figshare. https://doi.org/10.6084/m9.figshare.30204301 (2025).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Acknowledgements
We would like to express our gratitude to Kindstar Sequenon Co., Ltd for its support of the sequencing technology. This study was financially supported by the Project of the Science and Technology Program of Shaanxi Academy of Science (No. 2024k-32), National Natural Science Foundation of China (No. 32000256, 31900273), and the Innovation Capability Support Program of Shaanxi (No. 2025ZC-KJXX-119).
Author information
Authors and Affiliations
Contributions
X.Z. and Y.Z. conceived and designed the study. X.Z. and Y.Z. prepared the samples. X.Z., H.Z., and C.C. analyzed the data. X.Z. and H.Z. wrote the manuscript. C.C. and Y.Z. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Zhang, H., Chen, C. et al. A high-quality Chromosome-level genome assembly of Gynostemma guangxiense (Cucurbitaceae). Sci Data 13, 503 (2026). https://doi.org/10.1038/s41597-026-06889-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06889-x
