
Abstract
Effective diagnosis and treatment of lung adenocarcinoma depends on accurate typing, subtyping, and grading. Herein, we present the CLWD dataset, a valuable resource for the lung cancer pathology community, comprising 408 whole-slide images (WSIs) from 210 patients specifically curated for the study of lung adenocarcinoma subtypes. Scanned at 80 × magnification, it is one of the largest datasets in Asia, with a particular emphasis on Chinese patient demographics. Notably, the dataset includes comprehensive clinical information, such as age, sex, and diagnosis, providing a robust foundation for diverse research needs. Publicly accessible, it supports a range of applications, including machine learning model development and validation. An initial evaluation of lung adenocarcinoma subtype classification using a multi-instance learning framework demonstrated that this dataset can substantially advance global research and improve the accuracy of subtype diagnosis.
Similar content being viewed by others

Background & Summary
Lung cancer is the leading cause of cancer-related mortality worldwide, with an estimated 1.8 million deaths in 20201,2. Owing to China’s large population, the number of new lung cancer cases and deaths far exceeds that of any other country. Lung adenocarcinoma (LUAD), which accounts for approximately 50% of all lung cancer diagnoses3, represents the most prevalent subtype. LUAD exhibits distinct histologic and molecular characteristics, and its pronounced heterogeneity arises from the complex mixture of multiple coexisting growth patterns4. The 2015 World Health Organization (WHO) guidelines refined the classification of LUAD by emphasizing its predominant growth pattern and overall pattern of invasion3,4. LUADs that are predominantly lepidic and non-mucinous are classified according to their infiltration pattern as pre-invasive adenocarcinoma in situ (AIS), minimally invasive adenocarcinoma (MIA), or fully invasive adenocarcinoma (IA) with a lepidic component. In contrast, other invasive, non-adherent LUAD with distinct morphologic features are classified based on their predominant growth pattern5. Growing evidence indicates that different histologic subtypes of LUAD are closely associated with distinct risks of recurrence and overall prognosis6. Among these, the lepidic growth pattern is considered the least invasive, whereas the micropapillary and solid patterns are classified as the most invasive and are linked to higher recurrence rates and poorer survival outcomes. Meanwhile, the papillary and acinar growth patterns exhibit intermediate levels of invasiveness7. Early-stage LUADs are a heterogeneous group, and identifying IA subtypes with a high risk of recurrence, such as acinar, micropapillary, lepidic, solid, and cribriform patterns, is critical, as these patients may benefit from closer surveillance and potentially even adjuvant therapy8.
Genomic, transcriptomic, and proteomic analyses have become the focal point of cancer research in the past decades9,10. Meanwhile, histological phenotyping provides an additional key layer of information that complements genomic, transcriptomic, and proteomic data, and the integration of these modalities is essential for accurate cancer diagnosis11. Computational pathology refers to quantitative and algorithmic analysis of pathology images, including classical image analysis as well as machine-learning and deep-learning approaches, to extract clinically and biologically meaningful information from histologic phenotypes.
Histopathological analysis of tumor specimens involves several standardized procedures, including formalin fixation, tissue sectioning, paraffin embedding, and hematoxylin and eosin (H&E) staining, followed by microscopic examination by a trained pathologist. Using established diagnostic criteria, the pathologist determines the presence of tumor tissue, identifies the subtype, and evaluates key histological features. In addition, for many tumor types, quantitative assessments, such as tumor grading, evaluation of mitotic activity, quantification of tumor-infiltrating lymphocytes, and estimation of tumor burden, are routinely performed12. In clinical practice, microscopic examination of tissue slides is labor-intensive, time-consuming, and susceptible to inter-observer variability13. For example, in certain atypical breast cancer cases, the diagnostic concordance between pathologists can be as low as 48%14. Recent advances in scanning technology have enabled the digitization of slides into high-resolution images, facilitating the integration of digital pathology (DP) into routine pathological practice12. For instance, convolutional neural networks (CNNs) extend the capabilities of computer-based image analysis for many routine histopathology tasks, including tumor detection in breast, prostate, and esophageal cancers15,16. CNNs have also been applied to the subtype analysis of lung and kidney cancers17,18. A lightweight CNN, based on the AlexNet architecture, has been developed to assess the accuracy of identifying four LUAD subtypes, acinar, micropapillary, solid, and cribriform, in surgically resected specimens19. Moreover, DP enables the classification of cancers of unknown primary origin, which are often challenging to diagnose and treat20. The advancement of these technologies now makes it feasible to evaluate early-stage lung adenocarcinoma subtypes directly from DP images.
To support the need for diverse data in lung adenocarcinoma research, we present the Chinese Lung Adenocarcinoma WSI Dataset (CLWD), comprising digitized H&E whole-slide images (WSIs) from a Chinese patient cohort, each accompanied by detailed clinical information. Rigorous manual review ensures high image quality and reliable diagnostic accuracy. The dataset represents a valuable resource for digital and computational pathology, specifically curated to support the identification of lung adenocarcinoma subtypes, including acinar, cribriform, in situ, lepidic, micropapillary, papillary, and solid patterns. Figure 1 provides an overview of the data collection process and the deep learning pipeline. Otherwise, several publicly available LUAD histopathology resources have enabled method development and benchmarking. Notably, the Dartmouth Lung Cancer Histology Dataset (DHMC) provides de-identified LUAD whole-slide images with predominant-pattern annotations established by consensus review from three pathologists21. We include DHMC as a closely related reference cohort for comparative benchmarking against CLWD. Comprising 408 WSIs, the CLWD dataset aims to facilitate global collaboration in lung cancer research, expand the diversity of existing cohorts, and provide a foundation for the development of early diagnostic tools for lung adenocarcinoma subtypes.

Data collection and digitization workflow of CLWD. (A) LUAD tissue slides were collected and subjected to an initial quality control (QC) check, followed by digitization into WSIs and a second round of QC, and those passing QC proceeded to the next step. (B) A deep learning-based multi-instance learning (MIL) framework was applied to classify lung adenocarcinoma (LUAD) subtypes. A pre-trained neural network extracted patch-level features, which were aggregated into slide-level representations using a feature pooling approach. These aggregated features were then used by the TransMIL, CLAM, and Graph Transformer models for LUAD subtype classification. Abbreviations.
Methods
Data acquisition
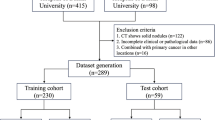
The present study retrospectively collected H&E-stained slides of various lung adenocarcinoma subtypes between 2020 and 2023. A total of 408 slides from 210 patients were obtained from hospital records. Of these, 80 slides were classified as in situ, 31 as cribriform, 30 as lepidic, 64 as micropapillary, 84 as papillary, 50 as acinar, and 69 as solid subtype (WHO 2015). We additionally provide a WHO 2021-compatible annotation layer. Specifically: (i) In the WHO 2021-compatible mode labels, the cribriform pattern has been merged into the acinar pattern, but cribriform remains as an optional morphological pattern in the original benchmarking field; (ii) Mucinous/non-mucinous status. The study was approved by the Ethics Committee of the First People’s Hospital of Yunnan Province (For a retrospective analysis of archival pathological materials and associated clinical metadata. The Ethics Committee of The First People’s Hospital of Yunnan Province approved the study with a waiver of informed consent because the research posed minimal risk to participants and used de-identified retrospective data. IRB Number: KHLL2025-KY126). To protect patient privacy, all identifying information was anonymized and replaced with unique sample identifiers. As illustrated in Figure 1A, the data collection and digitization workflow begins with tissue sectioning, followed by an initial quality control (QC) step. After digitization into WSI, a second round of QC is performed. Manual QC included microscope-based review of staining adequacy and tissue integrity, and WSI-level inspection of focus, color/stain uniformity, folds/bubbles/pen marks, and tissue completeness; slides failing QC were rescanned when feasible or excluded. For the classification of LUAD subtypes, a deep learning-based multiple instance learning (MIL) framework is employed (Figure 1B). A pre-trained neural network was employed to extract patch-level features, which were then aggregated into slide-level representations using a feature pooling method. These aggregated features were subsequently used to classify LUAD subtypes.
H&E-stained slides from all LUAD patients with histologic subtype labels were assigned based on routine diagnostic pathology review by board-certified lung cancer pathologists at our institution, following established LUAD predominant-pattern criteria. Immunohistochemistry was used when clinically indicated to support diagnosis. For each case, slides exhibiting the most representative histopathological features were selected for digitization, and cases considered diagnostically ambiguous for subtype assignment were excluded from the dataset during quality control. To ensure high-quality WSIs, representative sections were recut from the FFPE tissue blocks and restained prior to digitization. Slides were digitized using an SQ600P scanner at 80 × magnification, yielding a level of detail comparable to that of conventional microscopic observation. Manual quality control was performed to ensure high fidelity in the digital representations. Figure 2 illustrates WSIs of the various histologic subtypes of LUAD. The digitized images were exported and stored in .svs format.

Representative WSIs of different LUAD subtypes. (A) in situ subtype. (B) papillary subtype. (C) cribriform subtype. (D) solid subtype. (E) acinar subtype. (F) lepidic subtype. (G) micropapillary subtype.
Slide selection criteria
For each case, representative slides were selected by board-certified lung cancer pathologists to best reflect the LUAD subtypes used for dataset labeling. Representative slides were defined as those that: (i) contain sufficient viable tumor tissue with preserved architecture (avoiding extensive necrosis/hemorrhage/crush artefact); (ii) show clear pattern-defining structures (e.g., lepidic growth, acinar structures, papillary structures, micropapillary tufts, solid sheets formation); (iii) include diagnostically informative regions (e.g., invasive component when applicable); and (iv) exhibit adequate H&E staining quality and section integrity. When the initially available slide was suboptimal for digitization, representative slides were recut from FFPE blocks and restained prior to scanning.
Clinical data acquisition
Clinical information, including age, sex, and pathological subtype, was obtained from the pathological archives of the First People’s Hospital of Yunnan Province. The variables included in the database are summarized in Table 1. To protect patient privacy, all personal identifiers were removed, and data aggregation techniques were applied to ensure anonymization and prevent potential re-identification.
Data preprocessing
The preprocessing workflow begins with segmenting tissue regions in the digitized WSIs. Following image smoothing, the slides are converted to the HSV color space, and a mask is created by thresholding the saturation channel. Tissue regions of the WSI are then segmented into non-overlapping patches according to the specified magnification, with each patch measuring 256 × 256 pixels (All slides were digitized into WSIs with a resolution of 0.1 µm/pixel using the SQS-600P slide scanner from Guangzhou Shengqian Biotechnology Co., Ltd.). WSIs may contain occasional digitization artefacts (e.g., dust/hair, scanner-related striping, and large background margins). These artefacts are mainly observed in non-tissue background regions. Prior to patch extraction, we perform tissue detection to generate a tissue mask and exclude background/blank regions; only tissue-containing tiles are retained for feature extraction and MIL training/inference. Consequently, background artefacts (including striping and margins) are removed before model input.
Baseline models
This section describes the benchmark models used for the classification task on our dataset. Given the high resolution of histopathological images and the lack of detailed slide-level annotations, the models are implemented within a weakly supervised MIL framework. Unlike conventional supervised approaches that require precise instance-level labels (e.g., at the pixel or patch level), MIL treats each slide as a “bag” of instances, with labels assigned at the bag (slide) level based on the aggregate features of its constituent patches. This approach effectively transforms a fully supervised problem into a weakly supervised learning task. The MIL framework generally comprises two stages: (i) feature extraction and (ii) feature aggregation. To benchmark our dataset, we employed three classical classification networks commonly used in digital histopathology. These methods, briefly described below, are all based on self-attention mechanisms and are applied to classify LUAD WSIs into subtypes, including papillary, cribriform, in situ, solid, micropapillary, lepidic, and acinar. By evaluating these diverse approaches, we aim to comprehensively assess their generalization performance on the LUAD pathology dataset.
CLAM
CLAM (Clustering-constrained Attention Multiple Instance Learning) is a user-friendly, high-throughput, and interpretable approach for WSI-level analysis17. Operating under a weakly supervised paradigm, CLAM requires only slide-level labels while maintaining high data efficiency and adaptability. It is particularly well-suited for complex multi-class subtyping tasks. CLAM utilizes an attention-based deep learning framework to automatically identify diagnostically relevant subregions within whole-slide images, enabling accurate slide-level classification. It incorporates instance-level clustering on the representative regions selected by the attention mechanism. This clustering constraint refines the feature space, improving both model interpretability and classification performance.
TransMIL
To better capture the relational information among instances, TransMIL, a Transformer-based Multiple Instance Learning framework, was developed22. TransMIL leverages morphological and spatial features in WSIs, effectively addressing the inherent limitations of conventional MIL approaches, which often neglect structural relationships. By leveraging the self-attention mechanism of Transformers, TransMIL can model long-range dependencies across image patches, enabling more comprehensive feature interactions. This framework exhibits robust performance on both balanced and imbalanced datasets and supports binary as well as multi-class classification tasks. Experimental results demonstrate that TransMIL achieved higher classification accuracy and faster convergence compared with previous methods.
Graph Transformer
The Graph Transformer combines graph neural networks with Transformer mechanisms, introducing relational graph structures into the multiple instance learning framework23. It constructs a graph among image patches to model their spatial and semantic relationships. Using graph-based attention mechanisms, the model assigns adaptive weights to each node, allowing weighted aggregation and the generation of graph-level representations for slide-level multi-class classification. This architecture incorporates structural priors, enhancing the modeling of interactions among instances.
Experimentation setup
The employed deep learning models were implemented with high fidelity, closely following the official repositories to ensure methodological consistency. Before model training, a comprehensive evaluation of the dataset was performed to establish baseline performance metrics across all feature extraction and aggregation strategies. To rigorously prevent data leakage, we first perform a patient-level 9:1 split into a training set (90%) and an independent held-out test set (10%). Within the 90% training set, we then perform 5-fold cross-validation, splitting it into five equal folds: in each round, one fold is used as the validation set and the remaining four folds are used for training.
(To maintain robust results, in another experiment, patients were stratified and assigned exclusively to the training (80%), validation (10%), or test (10%) cohorts, ensuring that all image slices from a single subject remained within the same data partition.) Furthermore, all experiments were conducted using five-fold cross-validation to enhance the robustness and generalizability of the results. Model training was carried out for a minimum of 50 epochs and a maximum of 200 epochs, with early stopping applied if no improvement in validation loss was observed over 25 consecutive epochs. Aggregation modules were optimized using hyperparameters consistent with their original publications: TransMIL was trained with a learning rate of 2 × 10−4 and a weight decay of 5 × 10−³, CLAM used a learning rate of 2 × 10−⁴ and a weight decay of 1 × 10−³, and Graph Transformer employed a learning rate and weight decay of 1 × 10−⁴ each. All computational experiments were conducted on a server equipped with four Nvidia V100 32GB GPUs and an Intel Xeon Gold 6240 CPU, with 187 GB of RAM. Models were implemented in Python 3.8.20 using PyTorch 2.3.0. A full list of software dependencies and their versions is provided in the requirements.txt file in our GitHub repository.
Data Records
The dataset is publicly available via Figshare24 and can also be accessed directly through our Pathology Image Repository (https://leelab.kmmu.edu.cn/PathologyRepository). It contains high-resolution histopathological images intended to support researchers and clinicians in studying lung tumors, particularly LUAD subtypes. Key statistics from the CLWD dataset are summarized in Table 2. The repository facilitates a range of research applications, including machine learning model development and histopathological analysis. The dataset is organized as follows:
CWLD.csv
A detailed spreadsheet containing clinical information for each slide.
WSI_X.svs
H&E-stained WSIs in .svs format.
All WSIs in the dataset are labeled as “WSI-X,” where “X” represents a sequentially assigned de-identified patient identifier (e.g., WSI-1, WSI-2, WSI-3). This naming convention ensures patient anonymity. Corresponding clinical metadata for each WSI is provided in the “CLWD.csv” file. A comprehensive summary of the clinical information is presented in Tables 1,2.
To ensure the reliability and diagnostic utility of the dataset, a stringent quality assurance protocol was implemented. This process was overseen by board-certified lung cancer pathologists with extensive diagnostic experience. This protocol included (i) pre-scan microscopic inspection to confirm staining adequacy and representativeness, (ii) post-scan digital QC with rescanning when needed, and (iii) systematic exclusion of slides deemed diagnostically ambiguous or likely to cause subtype misinterpretation. This careful curation strategy maintains the integrity of the dataset and maximizes its suitability for downstream research applications. The JPG version of the dataset also available at the Hugging Face repository25 (https://huggingface.co/datasets/kmmuleelab/Lung_Pathology_Image_JPG).
To contextualize CLWD relative to existing public LUAD WSI resources, we additionally provide a comparison with the Dartmouth/DHMC LUAD dataset, including dataset characteristics and standardized baseline performance under matched evaluation protocols (Supplementary Table S1).
Technical Validation
Quality assurance procedures were applied at multiple stages of the data preparation pipeline. Before digitization, each histological slide was manually inspected under a light microscope to evaluate staining quality and ensure that the scanned images accurately represented the histopathological features critical for downstream analysis. After digitization, the WSIs were reviewed, and suboptimal images were either rescanned or excluded as needed. Notably, the dataset includes comprehensive clinical metadata, such as patient age, sex, diagnostic interpretation, and assessments of tissue integrity and accuracy. To validate the reliability of these annotations, a randomly selected subset was independently reviewed. This quality control process involved cross-referencing the original metadata with expert pathological assessments (performed by Y.C., L.W., R.S.L., and Y.H.J.) to confirm label accuracy. Such verification strengthens the dataset’s credibility and enhances its utility for developing and validating computational diagnostic models. This independent review was conducted as a label verification step for public release by cross-referencing the released metadata with expert slide interpretation. Because the full dataset labels primarily reflect finalized clinical sign-out diagnoses and we did not perform a fully crossed multi-rater design on all slides, we do not report inter-rater agreement statistics (e.g., Cohen’s kappa) in the current release, this is now explicitly stated as a limitation.
Performance metrics for baseline models
The primary evaluation metric across all experiments was the area under the receiver operating characteristic curve (AUC), chosen for its robustness and for providing an aggregate measure of model discrimination, particularly in the presence of class imbalance. Complementary metrics included slide-level accuracy (ACC), recall, precision, and F1 score, enabling a comprehensive assessment of classifier performance. Specifically, the AUC quantifies the model’s ability to distinguish between different classes. Precision measures the proportion of predicted positive instances that are true positives, assessing the reliability of positive predictions. Recall, in contrast, represents the proportion of actual positive cases correctly identified by the classifier, reflecting the model’s sensitivity. The F1 score, as the harmonic mean of precision and recall, provides a balanced metric, particularly useful for datasets with imbalanced class distributions, as it accounts for both false positives and false negatives. All models were evaluated using five-fold cross-validation (folds 0–4) to ensure robust performance assessment. For multi-class evaluation, we report macro-averaged metrics. Specifically, precision, recall, and F1 score are computed for each class and then averaged with equal weight across classes (macro averaging). To reporting baseline performance on CLWD, we benchmarked the same MIL pipelines on the publicly available Dartmouth/DHMC LUAD WSI dataset using identical preprocessing and evaluation protocols (the clinical information of DHMC dataset is presented in Supplementary Table S2). Comparative results across CLWD and DHMC are summarized in Supplementary Table S1.
LUAD subtype
Figure 3 and Supplementary Table S1 summarize the performance results of three established deep learning models for classifying lung adenocarcinoma subtypes using the CLWD dataset and DHMC dataset. Using a patient-level split strategy, we first reserved 10% of patients as an independent held-out test set and used the remaining 90% for model development, where a 5-fold rotation within the training set was used to select the validation fold for early stopping and model selection (one fold as validation, four folds as training). Figure 3 summarizes the mean one-vs-rest ROC curves (mean ± SD) and class-wise AUCs on the held-out test set for both cohorts. CLWD generally yielded stronger discrimination than the external DHMC cohort, indicating that the released CLWD WSIs and labels support robust learnability under standard MIL pipelines. In particular, on CLWD, all three baselines achieved high AUCs for in situ (CLAM: 94.02 ± 2.15; Graph Transformer: 88.08 ± 1.57; TransMIL: 96.16 ± 2.12) and maintained consistently competitive performance for multiple invasive patterns (e.g., lepidic, solid, and cribriform) (Fig. 3A,C). By contrast, performance on DHMC showed larger class- and model-dependent variability, with several classes exhibiting notably lower AUCs under the same pipelines (e.g., papillary and/or acinar for CLAM/Graph Transformer). While TransMIL performed strongly on DHMC - achieving high AUCs for solid (93.72 ± 2.19) and micropapillary (91.01 ± 11.43) (Fig. 3D,F) - the overall trend remained that CLWD provides equal or higher class-wise separability across most models and classes, supporting its utility as a high-quality LUAD WSI benchmark and a complementary resource for cross-cohort generalization studies.

Mean ROC curves under a patient-level 9:1 split with internal 5-fold validation. (A–C) CLWD cohort (7 classes). (D–F) DHMC cohort (5 classes). For each cohort, we reserved 10% of patients as an independent held-out test set and used the remaining 90% for model development, where the training set was partitioned into five equal folds and one fold was rotated as the validation set (four folds for training) for early model selection. Panels show mean one-vs-rest ROC curves for each class for (A,D) CLAM, (B,E) Graph Transformer, and (C,F) TransMIL. Shaded bands indicate variability across repeated runs/folds (mean ± SD). The dashed diagonal line denotes random performance. Class-wise AUC values are reported as mean ± SD on the held-out test set.
To further assess robustness under a fixed partitioning strategy, we conducted an additional experiment in which patients were stratified and assigned at the patient level to training (80%), validation (10%), and test (10%) cohorts, ensuring that all WSIs from a single patient remained within the same partition. This procedure was repeated five times (different random seeds), and results are summarized as mean ± SD of held-out test-set performance in Fig. 4. In particular, CLWD consistently yielded high discrimination for readily identifiable patterns (e.g., in situ and several invasive patterns) (Fig. 4A,C), whereas DHMC exhibited larger class-dependent fluctuations and reduced separability for certain patterns in some models (Fig. 4D,F). Although TransMIL remained comparatively robust on DHMC, the overall trend across models indicates that CLWD provides equal or superior test-set performance relative to DHMC under the stratified 8:1:1 setting, supporting CLWD as a strong benchmark resource and a complementary cohort for cross-dataset generalization analyses.

Baseline benchmarking under another stratified method (8:1:1). Panels show one-vs-rest ROC curves for the evaluated classes for (A,D) CLAM, (B,E) Graph Transformer, and (C,F) TransMIL on (A to C) CLWD dataset and (D to F) DHMC dataset. Shaded regions indicate variability across repeats (mean ± SD). The diagonal dashed line indicates chance-level performance.
To examine how label granularity affects model performance on CLWD, we conducted patient-level 5-fold cross-validation under two classification settings: a 6-class predominant-pattern (WHO 2021) task (acinar, lepidic, papillary, micropapillary, solid, and in situ) and a 3-class invasion-status task (AIS, MIA, IA). Supplementary Fig. S1 reports mean one-vs-rest ROC curves and class-wise AUCs (mean ± SD across folds).
In the 6-class setting, all baselines achieved strong discrimination for in situ and solid, while performance was comparatively lower for papillary and acinar, consistent with the known morphological overlap among intermediate-risk patterns. In the 3-class (AIS/MIA/IA), AIS and IA were consistently well separated across methods (AUCs > 87), whereas MIA remained the most challenging class, with substantial model-dependent variability (may be partially attributable to its smaller sample size, which can increase estimation variance and reduce model stability under cross-validation).
Overall, across these evaluation experiments, CLWD consistently supports strong and reproducible baseline learnability, highlighting its value as a high-quality LUAD WSI dataset and a practical foundation for future cross-cohort generalization studies and clinically relevant model development.
Usage Notes
All 408 WSIs in this dataset are stored in the standardized .svs format. Accompanying clinical metadata are provided in spreadsheet form and are accessible via Figshare. The dataset holds potential for integration with other publicly available resources to support deep learning model development and educational applications. Nonetheless, several limitations should be noted. All images were acquired at a single institution using a uniform scanning platform (SQS600P). Notably, class imbalance among LUAD subtypes may pose challenges for model training and affect generalizability. Moreover, individual WSIs may contain multiple histological subtypes, further complicating analysis. The absence of immunohistochemistry (IHC) marker-stained slides limits the dataset’s utility for analyses involving molecular biomarkers. Furthermore, because all images were acquired at a single institution using a uniform scanning platform, the resulting models may have reduced external validity. Additionally, LUAD subtypes are subject to inter-observer variability, while we performed expert QC and independent subset verification, formal inter-rater reliability metrics (e.g., Cohen’s kappa) across the full cohort are not provided in the current release. Researchers should consider these limitations when utilizing the dataset.
Data availability
The dataset is publicly available via Figshare24 and can also be accessed directly through our Pathology Image Repository (https://leelab.kmmu.edu.cn/PathologyRepository). Otherwise, the JPG version of the dataset also available at the Hugging Face repository (https://huggingface.co/datasets/kmmuleelab/Lung_Pathology_Image_JPG).
Code availability
The code for preprocessing and deep learning models is publicly available on GitHub: https://github.com/DrNeilChen/CLWD.
References
Lortet-Tieulent, J. et al. International trends in lung cancer incidence by histological subtype: adenocarcinoma stabilizing in men but still increasing in women. Lung Cancer 84, 13–22 (2014).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71, 209–249 (2021).
Travis, W. D. et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol 10, 1243–1260 (2015).
Travis, W. D. et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 6, 244–285 (2011).
Xiang, C. et al. Distinct mutational features across preinvasive and invasive subtypes identified through comprehensive profiling of surgically resected lung adenocarcinoma. Mod Pathol 35, 1181–1192 (2022).
Caso, R. et al. The Underlying Tumor Genomics of Predominant Histologic Subtypes in Lung Adenocarcinoma. J Thorac Oncol 15, 1844–1856 (2020).
Zhang, Y. et al. Excellent Prognosis of Patients With Invasive Lung Adenocarcinomas During Surgery Misdiagnosed as Atypical Adenomatous Hyperplasia, Adenocarcinoma In Situ, or Minimally Invasive Adenocarcinoma by Frozen Section. Chest 159, 1265–1272 (2021).
Zhai, W. et al. Prognostic Nomograms Based on Ground Glass Opacity and Subtype of Lung Adenocarcinoma for Patients with Pathological Stage IA Lung Adenocarcinoma. Front Cell Dev Biol 9, 769881 (2021).
Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Chen, F. et al. Moving pan-cancer studies from basic research toward the clinic. Nat Cancer 2, 879–890 (2021).
Shmatko, A., Ghaffari Laleh, N., Gerstung, M. & Kather, J. N. Artificial intelligence in histopathology: enhancing cancer research and clinical oncology. Nat Cancer 3, 1026–1038 (2022).
Janowczyk, A. & Madabhushi, A. Deep learning for digital pathology image analysis: A comprehensive tutorial with selected use cases. J Pathol Inform 7, 29 (2016).
Ozkan, T. A. et al. Interobserver variability in Gleason histological grading of prostate cancer. Scand J Urol 50, 420–424 (2016).
Elmore, J. G. et al. Diagnostic concordance among pathologists interpreting breast biopsy specimens. JAMA 313, 1122–1132 (2015).
Campanella, G. et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat Med 25, 1301–1309 (2019).
Gehrung, M. et al. Triage-driven diagnosis of Barrett’s esophagus for early detection of esophageal adenocarcinoma using deep learning. Nat Med 27, 833–841 (2021).
Lu, M. Y. et al. Data-efficient and weakly supervised computational pathology on whole-slide images. Nat Biomed Eng 5, 555–570 (2021).
Yang, H. et al. Deep learning-based six-type classifier for lung cancer and mimics from histopathological whole slide images: a retrospective study. BMC Med 19, 80 (2021).
Gertych, A. et al. Convolutional neural networks can accurately distinguish four histologic growth patterns of lung adenocarcinoma in digital slides. Sci Rep 9, 1483 (2019).
Lu, M. Y. et al. AI-based pathology predicts origins for cancers of unknown primary. Nature 594, 106–110 (2021).
Wei, J. W. et al. Pathologist-level classification of histologic patterns on resected lung adenocarcinoma slides with deep neural networks. Sci Rep 9, 3358 (2019).
Shao, Z. et al. TransMIL: Transformer based Correlated Multiple Instance Learning for Whole Slide Image Classication. in Neural Information Processing Systems (2021).
Zheng, Y. et al. A Graph-Transformer for Whole Slide Image Classification. IEEE Trans Med Imaging 41, 3003–3015 (2022).
Chen, Y. CLWD: a Chinese histopathology dataset for lung adenocarcinoma subtype classification. figshare https://doi.org/10.6084/m9.figshare.29035847 (2025).
Chen, Y. et al. Lung_Pathology_Image_JPG (Revision 312c831). Hugging Face https://doi.org/10.57967/hf/7794 (2026).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 82560572, No. 82404091, and No. 62302429), the Yunnan Province Applied Basic Research Program Kunming Medical University Joint Project (202401AY070001-120), the Health Commission Foundation of Yunnan Province (2023-KHRCBZ-B15), the Kunming University of Science and Technology Joint Medical Project (KUST-KH2023013Y), Yunnan Fundamental Research Projects(202501CF070023), Kunming University of Science and Technology Joint Medical Project (KUST-KH2022018Y), Major Science and Technology Projects of Yunnan Province (202402AA310016), Basic Research Science and Technology Foundation of Yunnan Province (202201AS070009) and Xing Dian Foundation of Yunnan Province (XDYC-MY-2022-0029).
Author information
Authors and Affiliations
Contributions
Conceptualization: J.P., J.L., D.P.T. and J.N.; Methodology and formal analysis: Y.C., H.Y.Z. and J.L.; Investigation: Y.C., H.Y.Z. and L.W.; Data curation: L.W., L.L., R.S.L., Y.H.J., P.R.T. and Y.L.; Writing-Original Draft: Y.C. and H.Y.Z.; Writing-Review & Editing: L.L., J.P., J.L., D.P.T. and J.N.; Supervision: J.P., J.L., D.P.T. and J.N. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Y., Zhao, H., Wang, L. et al. CLWD: a Chinese histopathology dataset for lung adenocarcinoma subtype classification. Sci Data 13, 599 (2026). https://doi.org/10.1038/s41597-026-06906-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06906-z
