
Abstract
Species of the genus Varanus exhibit substantial variation in body size, making them an excellent model system for studying evolutionary biology. Their genomes can provide valuable insights into the evolutionary mechanisms underlying body size diversity in vertebrates. Here, we presented a chromosome-level genome assembly for the water monitor lizard (Varanus salvator), generated and annotated through an integrated multi-omics approach. The assembled genome spans 1,645 Mb, with contig and scaffold N50 values of 27.34 Mb and 12.63 Mb, respectively. Approximately 97.4% of the assembled sequences were anchored onto 20 pseudochromosomes using Hi-C contact data. Repetitive elements accounted for approximately 34.0% of the genome. Assembly completeness was assessed with BUSCO, revealing that 96.7% of the conserved vertebrate BUSCO genes were complete. We identified 19,347 protein-coding genes by integrating evidence from three complementary approaches. Among these, 98.8% were functionally annotated using at least one of six major protein databases. This high-quality, chromosomal-level genome provides a critical resource for future studies in reptilian biology, encompassing evolution, ecological adaptation, and conservation.
Similar content being viewed by others

Background & Summary
Monitor lizards (family Varanidae) are represented by a single genus, Varanus, which comprises 88 currently recognized species1. Many of these species have only recently been recognized as distinct, based primarily on genetic evidence. Furthermore, the number of newly identified cryptic species—morphologically similar or nearly identical forms—is steadily increasing2. Varanid lizards exhibit remarkable body size disparity, ranging from the diminutive Dampier Peninsula monitor (V. sparnus; approximately 230 mm in length and <17 g) to the massive Komodo dragon (V. komodoensis; up to 3,000 mm in length and 100 kg)2. Distributed primarily across eastern Africa and southern Australia, varanid lizards also inhabit diverse island systems (including oceanic, land-bridge, and continental fragments) throughout New Guinea, the Philippines, Indonesia, and the Solomon Islands. This distribution renders them an excellent model system for studying the roles of continents and islands in species evolution and diversification3. Additionally, many varanid species are involved in international trade and are listed under the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES)2. Consequently, monitor lizards represent a taxon of significant scientific value for studying continental and insular drivers of species differentiation, as well as considerable conservation importance. However, to date, chromosome-level genome assemblies have been published only for the ridgetail monitor (V. acanthurus)4, while scaffold-level genome assemblies are available for just five other Varanus species: V. komodoensis5,6, and the Southeast Asian water monitor (V. salvator macromaculatus)7, the Nile monitor (V. niloticus)8, the white-throated monitor (V. albigularis)8, and the blue tree monitor (V. macraei)9. Therefore, generating high-quality genomic resources for additional varanid species remains a priority.
The water monitor lizard (V. salvator; Fig. 1a), which can reach up to 1,170 mm in snout-vent length10, is oviparous species. It is listed in Appendix II of the Convention on the International Trade of Endangered Species (CITES) and is a Class I key protected wild animal in China (https://www.gov.cn/zhengce/2021-02/05/content_5727412.htm). Its distribution extends from South and Southwest China across Bangladesh, Brunei, Indochinese Peninsula, northeastern India, Indonesia, and Sri Lanka11 (Fig. 1b). Water monitor lizards typically inhabit areas close to water sources, such as rivers, lakes, and marshes, and are highly adapted to both aquatic and terrestrial environments2. Like other ectothermic vertebrates, they rely on external heat sources to regulate their body temperature and exhibit a strong adaptive capacity to adapt to environmental fluctuations12. They are carnivorous, with a diet consisting primarily of snails, crabs, fish, frogs, other small vertebrates, and carrion13,14. This species plays an important role in maintaining ecological balance through regulating prey populations and facilitating nutrient cycling15,16.

Photo (a) and potential habitat distribution area (b) of the water monitor lizard (Varanus salvator). The species distribution was projected using maximum entropy (MaxEnt) modeling based on 276 occurrence records, following Guo et al.63.
Varanus salvator is critically endangered in China17 and holds cultural significance, having been traditionally regarded as the “five-clawed golden dragon”, a symbol of imperial power in Chinese culture18. Karyotype analysis has established its diploid chromosome number as 2n = 4019, and genome survey analysis estimates its total genome size at approximately 1.67 Gb. In this study, we generated a chromosome-level genome assembly for V. salvator by integrating data from Illumina short-read sequencing (104× coverage), PacBio single-molecule real-time (SMRT) long-read sequencing (105×), 10× Genomics linked-read sequencing (111×), and high-throughput chromosome conformation capture (Hi-C) (102×) sequencing. K-mer-based analysis estimated the assembled genome size to be 1.64 Gb, with a contig N50 length of 27.34 Mb and a GC content of 44.2% (Fig. 2a; Table 1). Approximately 97.4% of the assembled sequences were anchored onto 20 pseudochromosomes (Fig. 2b). Comparative genomic analysis identified the 16th chromosome as the Z sex chromosome (Fig. 2b). The genome exhibits a heterozygosity rate of 0.24% and contains 34.0% repetitive sequences, comprising LTR retrotransposons (6.96%, 114,420 Mb), LINE retrotransposons (28.8%, 472,895 Mb), and other types (Fig. 2a; Table 2). A total of 19,347 protein-coding genes were identified, with an average coding sequence (CDS) length of 1,572 bp. Of these, 19,116 (98.8%) were functionally annotated (Table 3). In addition, 3,275 non-coding RNAs were identified, comprising 719 miRNAs, 984 tRNAs, 432 rRNAs, and 369 snRNAs (Table 2). Assessments of genome completeness and continuity yielded high-quality metrics, with BUSCO and CEGMA scores of 96.7% and 87.9%, respectively. This high-quality, chromosome-level genome assembly and its annotation for V. salvator provide a valuable resource for ecological and evolutionary studies within the genus Varanus and lay a foundation for future research in molecular ecology and conservation genetics.

Genomic features and chromosomal assignment of V. salvator. (a) Circos plot of genome characteristics, showing from the outside to the inside chromosome ideograms, gene density, GC content, and collinearity block of self-vs-self. See Tables 1–3 for more detailed statistics. (b) Identification of the Z sex chromosome (Chromosome 16) in V. salvator by comparing with V. acanthurus.
Methods
Sample collection and genomic sequencing
An adult male water monitor lizard was sourced from Hainan Key Laboratory for Herpetological Research. Following its natural death, tissues including skin, fat bodies, liver, testis, spleen, muscle, pancreas, and other organs were collected and stored separately at −80 °C for subsequent analysis. All procedures were conducted in accordance with prevailing Chinese regulations on animal welfare and scientific research and were approved by the Animal Research Ethics Committee of Nanjing Normal University (IACUC Approval No. 20200511). Genomic DNA was extracted from muscle tissue using a standard phenol-chloroform-isoamyl alcohol (PCI; 25:24:1, v/v/v) method, followed by precipitation with chloroform-isoamyl alcohol (24:1, v/v). DNA concentration was quantified using a Qubit 2.0 fluorometer (Thermo Fisher Scientific). DNA purity and integrity were assessed using a Nanodrop spectrophotometer (Thermo Fisher Scientific) and by 1.0% agarose gel electrophoresis, respectively.
Whole-genome sequencing was performed using a combination of four platforms: Illumina short-read, PacBio single-molecule real-time (SMRT) long-read, 10× Genomics linked-read, and Hi-C technologies. For genome survey analysis, a paired-end library with an insert size of 350 bp was constructed and sequenced on an Illumina HiSeq X Ten System (Novogene, Beijing, China). After adapter trimming and removal of low-quality reads, more than 99.8% of the sequences were retained as clean data, yielding approximately 303 Gb. K-mer frequency analysis was performed on the paired-end short reads using Jellyfish v2.2.620 with a k-mer size of 21 (Fig. 3a). The resulting k-mer distribution was analyzed with GenomeScope v1.021 to estimate genome size, heterozygosity, and repetitive sequence content.

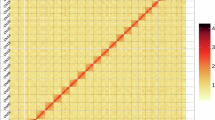
Genome survey of the Water Monitor Lizard (Varanus salvator) and chromosomes assembly, showing k-mer-21-spectra output generated by GenomeScope2 using PacBio HiFi reads and the estimated genome size and heterozygosity rate (a), and the Hi-C heatmap for chromosome interactions among the chromosomes of V. salvator (b).
Genomic DNA was randomly fragmented, and the resulting fragments were size-selected and purified using AMPure PB beads (Pacific Biosciences, USA). After end-repair and A-tailing, SMRTbell hairpin adapters were ligated for library construction. To maximize read length, libraries with insert sizes greater than 15 kb were size-selected using the BluePippin System (Sage Science, USA). These libraries were sequenced on a PacBio Sequel II system (Novogene) in continuous long read (CLR) mode, producing long reads with a random error profile. Sequencing yielded 175.33 G of raw data, corresponding to approximately 105× genome coverage. These long, error-prone reads are suitable for assembly using overlap-layout-consensus algorithms.
A 10× Genomics library was prepared using the Chromium Controller instrument and a Chromium Genome Chip following the manufacturer’s protocol for the Chromium Genome Reagent Kit (10× Genomics)22. The library was sequenced on an Illumina HiSeq X Ten platform. After quality control, 185.09 Gb of clean data were obtained, achieving approximately 111× genome coverage.
A Hi-C library was constructed from muscle tissue following a previously published protocol23 with minor modifications. Cells were cross-linked with 4% formaldehyde. The cross-linked DNA was then extracted and digested with 400 units of the restriction enzyme MboI on a rocking platform. The digested fragments were end-repaired and labeled with biotin-14-dCTP, followed by blunt-end ligation. Following ligation, the DNA was purified, fragmented into approximately 300–500 bp pieces, and used to construct Illumina-compatible paired-end sequencing libraries. The libraries were PCR-amplified (12–14 cycles) and sequenced on an Illumina Novaseq 6000 platform (PE 125 bp). This yielded 170.85 Gb of clean data, providing approximately 102× genome coverage.
Genome assembly and quality assessment
The initial de novo assembly was performed with Falcon v0.5, which implements an overlapping-layout-consensus (OLC) algorithm for assembling PacBio long reads24. Key overlap filtering parameters were set as follows:–max_diff 100–max_cov 100–min_cov 2–bestn 10, and the analysis was run using–n_core 12 computational threads. Error correction of long reads was performed using the Illumina short reads. Specifically, Illumina short reads were mapped to the PacBio long reads for error correction, leveraging the high base accuracy of short reads to polish the long-read sequences. This process yielded corrected long reads with high accuracy. These corrected reads were then assembled into primary consensus contigs by the OLC algorithm in Falcon. The primary assembly was further polished using Pilon v1.2325. Pilon polishing used the aligned Illumina short reads and was executed with default parameters as recommended, generating a polished assembly.
Subsequently, linked reads from the 10× Genomics libraries were aligned to the polished assembly. Scaffolds were constructed from the polished contigs using fragScaff v140324.126 based on these alignments. The scaffolding process was executed with the following parameters: -maxCore 200 for thread allocation; -fs1 -m 3000 -1 30 -E 25000 -o 50000 to define insert size, mapping quality, fragment length, and overlap thresholds for valid read pairs; advanced options -fs2′-C 3′ and -fs3′-j 1 -u 2′. For chromosome-scale scaffolding, we used the LACHESIS tool27 with Hi-C data. Hi-C reads were mapped to the scaffolded assembly. Using LACHESIS, contigs were then clustered, ordered, and oriented into chromosome-scale scaffolds with parameters optimized for our data: CLUSTER_N = 17, CLUSTER_MIN_RE_SITES = 2076, CLUSTER_MAX_LINK_DENSITY = 3, and CLUSTER_DRAW_HEATMAP = CLUSTER_DRAW_DOTPLOT = 1. Other parameters were set as default values. The resulting interaction matrices were used to validate the assembly, correct potential misassemblies, and ultimately cluster, order, and orient the scaffolds into pseudochromosomes (Fig. 3b). Thus, the final chromosome-level genome assembly was generated through the integration of data from all four aforementioned sequencing technologies.
To identify sex chromosomes, we performed a whole-genome alignment between the V. salvator assembly and the published V. acanthurus genome using MUMmer v3.028. The comparative results were visualized using Circos software.
Gene prediction and functional annotation
Repetitive elements were annotated using a combination of homology-based and de novo prediction approaches. First, for homology-based prediction, RepeatMasker v4.0.529 and ProteinMask v4.0.5 were run with default parameters against the Repbase database (v2018-10-26)30 to identify known repeats. Tandem repeats were predicted de novo using Tandem Repeats Finder (TRF) v4.0931 with default parameters. Next, for de novo repeat annotation, LTR_Finder v1.0.732, RepeatScout v1.0.533, and RepeatModeler v1.0.834 were used with default parameters to construct a de novo repeat library. Sequences longer than 100 bp with less than 5% ambiguous bases (‘N’s) were curated from the de novo predictions to generate a custom transposable element (TE) library. A non-redundant combined repeat library was created by merging the Repbase and custom TE libraries. Finally, RepeatMasker was then run against this combined library using ProteinMask v4.0.5 as the search engine to perform comprehensive repeat masking and annotation.
Protein-coding gene structures were predicted by integrating evidence from three approaches: homology-based, ab initio, and transcriptome-based. For homology-based prediction, protein sequences from six reptilian species (Sphenodon punctatus, Ophiosaurus gracilis, Anolis carolinensis, Shinisaurus crocodilurus, Pogona vitticeps, and Gekko japonicus) were aligned to the genome using TBLASTN v2.2.26 (E-value ≤ 10−5)35. Significant hits were extended into full-length gene models using GeneWise v2.4.136. For ab initio prediction, we used Augustus v3.2.337, GeneID v1.438, GeneScan v1.039, GlimmerHWM v3.0440, and SNAP (v2013-11-29)41. For transcriptome-based annotation, total RNA was extracted from seven tissues (skin, fat body, liver, testis, kidney, spleen, muscle, and pancreas) of the same specimen. RNA integrity and concentration were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Strand-specific RNA-seq libraries were prepared and sequenced on an Illumina NovaSeq 6000 platform (2 × 150 bp paired-end), yielding approximately 23 Gb of clean data. After adapter trimming and quality filtering (following the same procedure as for genomic DNA libraries), the transcriptome reads were de novo assembled using Trinity v2.1.142. The RNA-seq reads from each tissue were aligned to the genome assembly using TopHat v2.0.1143 for the identification of exon boundaries and splice junctions. Corresponding transcript assemblies were generated from the alignments using Cufflinks v2.2.144 with default parameters. Predictions from the three approaches were integrated using EvidenceModeler (EVM) v1.1.145 to generate a consensus, non-redundant set of gene models. The EVM gene models were further refined using the Program to Assemble Spliced Alignment (PASA), which leverages transcriptome assembly evidence to add untranslated regions (UTRs), identify alternative splicing isoforms, and produce the final gene set.
Subsequently, gene functions were annotated by searching predicted protein sequences against public databases. Predicted protein sequences were aligned against the Swiss-Prot database46 using BLASTP with an E-value ≤ 10−5, and the best significant hit was used for functional annotation. Protein domains and motifs were identified using InterProScan v4.847, which interrogates multiple databases including ProDom48, PRINTS49, Pfam50, SMART51, PANTHER52, and PROSITE53. Gene Ontology (GO) terms were assigned based on the InterProScan results. Additional functional annotations were assigned based on the best BLAST hits (E-value < 10−5) against the NCBI non-redundant (NR) and Swiss-Prot databases. Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) terms and pathway mappings were assigned using the KEGG Automatic Annotation Server (KAAS)54. tRNAs were identified using tRNAscan-SE v2.055. rRNAs were identified by aligning known rRNA sequences from related species to the assembly using BLASTN. Other non-coding RNAs (e.g., miRNAs, snRNAs) were identified by searching against the Rfam database v14.4 using Infernal v1.1.3 with default parameters.
Data Records
The genomic datasets generated and analysed in this study are available at the following repositories. All raw sequencing data [including Illumina short reads (CRR2192420-23), PacBio long reads (CRR2192408-10), 10× Genomics linked reads (CRR2192416-19), Hi-C data (CRR2192411-15), and RNA-seq data from skin (CRR2192430), fat body (CRR2192424), liver (CRR2192426), testis (CRR2192429), kidney (CRR2192425), spleen (CRR2192428), muscle (CRR2192431), and pancreas (CRR2192427)] and the primary genome assembly were deposited at the National Genomics Data Center (NGDC; https://ngdc.cncb.ac.cn/)56,57 under BioProject accession number PRJCA04577358. The final chromosome-scale genome assembly and annotation files are available at the NGDC Genome Warehouse (GWH) under accession number GWHHKDC00000000.159.
Technical Validation
The quality and completeness of the V. salvator genome assembly were assessed through multiple complementary approaches. First, analysis of the GC content distribution indicated no significant contamination in the assembly (Fig. 2a). Second, the Hi-C contact map revealed strong intra-chromosomal interaction signals along the diagonal (Fig. 3b), confirming the structural integrity of the genome. Assembly completeness was assessed using Benchmarking Universal Single-Copy Orthologues (BUSCO) v5.0 with the vertebrata_odb10 database (v2020-09-10), which contains 978 conserved single-copy orthologs60. The analysis showed that 96.7% of the expected single-copy orthologs were complete (C: 96.7%, of which 95.7% were single-copy and 1.0% duplicated; F: 0.8% fragmented; M: 2.5% missing; n: 978). In addition, completeness was evaluated using CEGMA v2.561 based on 248 highly conserved eukaryotic genes, which showed that 87.9% of the core genes were successfully assembled. To assess assembly accuracy, Illumina short reads were aligned to the final genome assembly using BWA v0.7.1762, resulting in a mapping rate of 99.4% and a genome coverage of 99.7%. Furthermore, 19,347 (98.8%) of the predicted gene models were functionally annotated against major databases, including Swiss-Prot, NR, KEGG, GO, Pfam, and InterPro. Collectively, these results indicate that our de novo assembly of the V. salvator genome is both high-quality and complete.
Data availability
All raw sequencing data generated in this study (Illumina short reads, PacBio long reads, 10× Genomics linked reads, Hi-C data, and RNA-seq data from skin, fat body, liver, testis, kidney, spleen, muscle, and pancreas) have been deposited at NGDC GSA (PRJCA045773, CRR2192408–31) and assembled chromosome-level genome at NGDC GWH (GWHHKDC00000000.1).
Code availability
No custom scripts or software were generated for this study. All data analyses were performed using published bioinformatics software, primarily using default parameters and according to the protocols described in the respective publications, unless otherwise specified.
References
Uetz, P., Freed, P. & Hošek, J. The Reptile Database, http://www.reptile-database.org, accessed on 21 June 2025. (2025).
Auliya, M. & Koch, A. Visual identification guide for the monitor lizard species of the world (genus Varanus). (Bundesamt für Naturschutz, Germany, 2020).
Zhu, X.-M. et al. The geographical diversification in varanid lizards: the role of mainland versus island in driving species evolution. Current Zoology 66, 165–171, https://doi.org/10.1093/cz/zoaa002 (2020).
Zhu, Z.-X., Dobry, J., Wapstra, E., Zhou, Q. & Ezaz, T. Gene traffic mediated by transposable elements shaped the dynamic evolution of ancient sex chromosomes of varanid lizard. Journal of Genetics and Genomics 3, 11, https://doi.org/10.1016/j.jgg.2025.08.002 (2025).
Lind, A. L. et al. Genome of the komodo dragon reveals adaptations in the cardiovascular and chemosensory systems of monitor lizards. Nature Ecology & Evolution 3, 1241–1252, https://doi.org/10.1038/s41559-019-0945-8 (2019).
Edward Via College of Osteopathic Medicine. Model organism or animal sample from Varanus komodoensis. GenBank https://identifiers.org/insdc.gca:GCA_007859595.1 (2025).
Chetruengchai, W. et al. Genome of Varanus salvator macromaculatus (Asian water monitor) reveals adaptations in the blood coagulation and innate immune system. Frontiers in Ecology and Evolution 10, 850817, https://doi.org/10.3389/fevo.2022.850817 (2022).
Colston, T. J., Pirro, S. & Pyron, R. A. The complete genome sequences of 101 species of reptiles. Biodiversity Genomes https://doi.org/10.56179/001c.129597 (2025).
Iridian Genomes. Model organism or animal sample from Varanus macraei. GenBank https://identifiers.org/insdc.gca:GCA_047404615.1 (2025).
Traeholt, C. Population dynamics and status of the water monitor lizard Varanus salvator in two different habitats in Malaysia. Wetlands International 1998, 147–160 (1998).
Du, Y., Lin, L.-H., Yao, Y.-T., Lin, C.-X. & Ji, X. Body size and reproductive tactics in varanid lizards. Asian Herpetological Research 5, 263–270, https://doi.org/10.3724/SP.J.1245.2014.00263 (2014).
Gleeson, T. T. Preferred body temperature, aerobic scope, and activity capacity in the monitor lizard, Varanus salvator. Physiological Zoology 54, 423–429, https://doi.org/10.1086/physzool.54.4.30155835 (1981).
Twining, P. J. & Koch, A. Dietary notes and foraging ecology of south-east Asian water monitors (Varanus salvator) in Sabah, northern Borneo, Malaysia. Herpetological Bulletin 143, 36–38 (2018).
Yang, J.-H. & Chan, B. P. L. Distribution, status, and ecology of the water monitor (Varanus salvator) on Hainan Island, and the role of folklore in its conservation. Herpetological Conservation and Biology 15, 427–439 (2020).
Moleón, M., Sánchez-Zapata, J. A., Selva, N., Donázar, J. A. & Owen-Smith, N. Inter-specific interactions linking predation and scavenging in terrestrial vertebrate assemblages. Biological Reviews 89, 1042–1054, https://doi.org/10.1111/brv.12097 (2014).
Twining, J. P., Bernard, H. & Ewers, R. M. Increasing land-use intensity reverses the relative occupancy of two quadrupedal scavengers. PLoS One 12, e0177143, https://doi.org/10.1371/journal.pone.0177143 (2017).
Wang, Y.-Z. Reptiles (I). China’s red list of biodiversity: vertebrates vol. III (Science Press, Beijing, 2021).
Shi, B.-Z. Zoological specimen museum catalogue in Changzhi College. (Shaanxi Science & Technology Press, Xi’an, 2018).
Srikulnath, K., Uno, Y., Nishida, C. & Matsuda, Y. Karyotype evolution in monitor lizards: Cross-species chromosome mapping of cDNA reveals highly conserved synteny and gene order in the Toxicofera clade. Chromosome Research 21, 805–819, https://doi.org/10.1007/s10577-013-9398-0 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M. & Jaffe, D. B. Direct determination of diploid genome sequences. Genome Research 27, 757–767, https://doi.org/10.1101/gr.214874.116 (2017).
Belton, J.-M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nature Methods 13, 1050–1054, https://doi.org/10.1038/nmeth.4035 (2016).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Mostovoy, Y. et al. A hybrid approach for de novo human genome sequence assembly and phasing. Nature Methods 13, 587–590, https://doi.org/10.1038/nmeth.3865 (2016).
Bickhart, D. M. et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nature Genetics 49, 643–650, https://doi.org/10.1038/ng.3802 (2017).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biology 5, R12, https://doi.org/10.1186/gb-2004-5-2-r12 (2004).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0. 2013-2015, http://www.repeatmasker.org. (2022).
Bao, W.-D., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Smit, A. & Hubley, R. RepeatModeler Open-1.0, http://www.repeatmasker.org/RepeatModeler/ 2008, accessed February 18 2019. (2019)
Johnson, M. et al. NCBI BLAST: a better web interface. Nucleic Acids Research 36, W5–W9, https://doi.org/10.1093/nar/gkn201 (2008).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Research 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Parra, G., Blanco, E. & Guigó, R. GeneID in Drosophila. Genome Research 10, 511–515, https://doi.org/10.1101/gr.10.4.511 (2000).
Lynn, A. M. et al. An automated annotation tool for genomic DNA sequences using GeneScan and BLAST. Journal of Genetics 80, 9–16, https://doi.org/10.1007/BF02811413 (2001).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nature Biotechnology 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111, https://doi.org/10.1093/bioinformatics/btp120 (2009).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols 7, 562–578, https://doi.org/10.1038/nprot.2012.016 (2012).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its new supplement TREMBL. Nucleic Acids Research 24, 21–25, https://doi.org/10.1093/nar/24.1.21 (1996).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Research 33, W116–W120, https://doi.org/10.1093/nar/gki442 (2005).
Corpet, F., Gouzy, J. & Kahn, D. The ProDom database of protein domain families. Nucleic Acids Research 26, 323–326, https://doi.org/10.1093/nar/26.1.323 (1998).
Attwood, T. K. et al. PRINTS-S: the database formerly known as PRINTS. Nucleic Acids Research 28, 225–227, https://doi.org/10.1093/nar/28.1.225 (2000).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res 49, D412–D419, https://doi.org/10.1093/nar/gkaa913 (2021).
Schultz, J., Copley, R. R., Doerks, T., Ponting, C. P. & Bork, P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Research 28, 231–234, https://doi.org/10.1093/nar/28.1.231 (2000).
Thomas, P. D. et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Research 31, 334–341, https://doi.org/10.1093/nar/gkg115 (2003).
Sigrist, C. J. A. et al. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Research 38, D161–D166, https://doi.org/10.1093/nar/gkp885 (2010).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene prediction. Methods in molecular biology, vol 1962 (ed. Kollmar, M.) 1–14 https://doi.org/10.1007/978-1-4939-9173-0_1 (Humana, New York, 2019).
CNCB-NGDC Members and Partners. Database resources of the national genomics data center, China National Center for Bioinformation in 2025. Nucleic Acids Research 53, D30–D44, https://doi.org/10.1093/nar/gkae978 (2025).
Chen, T.-T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics, Proteomics & Bioinformatics 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/search/all?q=PRJCA045773 (2026).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/search/all?q=GWHHKDC00000000.1 (2025).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Molecular Biology and Evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Parra, G., Bradnam, K., Ning, Z.-M., Keane, T. & Korf, I. Assessing the gene space in draft genomes. Nucleic Acids Research 37, 289–297, https://doi.org/10.1093/nar/gkn916 (2009).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Guo, K. et al. Climate warming will increase chances of hybridization and introgression between two Takydromus lizards (Lacertidae). Ecology and Evolution 11, 8576–8584, https://doi.org/10.1002/ece3.7671 (2021).
Acknowledgements
The study was supported by grants from the Scientific Research Foundation of Hainan Tropical Ocean University (RHDRCZK202535), the Technology Project of Hainan Province (ZDYF2018219), the Provincial Key Specialized Discipline Construction Project of Marine Science in Hainan Province (Hainan Education Department Higher Education Policy [2017], No. 153), and the National Key Program of Research and Development, Ministry of Science and Technology of China (2023YFF1304800). The authors would like to thank Jia-Tian Chen, Zhong-Yin Chen, Jian-Chao Fu, Cheng-Wang Li, and Qi-Ze Liu for help in sample collection during the research.
Author information
Authors and Affiliations
Contributions
Yu Du and Xia-Ming Zhu carried out the formal data analysis and drafted the manuscript under the supervision of Yan-Fu Qu and Xiang Ji. Yu Du, Xia-Ming Zhu, Yun-Tao Yao, Li-Ping Kang, Chi-Xian Lin, Xi-Feng Wang, and Kun Guo collected samples and conducted the laboratory experiments. Yu Du, Xia-Ming Zhu, Yan-Fu Qu, and Xiang Ji wrote the manuscript. All authors contributed to the review process and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Du, Y., Zhu, XM., Yao, YT. et al. Chromosome-scale genome assembly and annotation of the water monitor lizard, Varanus salvator. Sci Data 13, 594 (2026). https://doi.org/10.1038/s41597-026-06985-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06985-y
