
Abstract
Neoseiulus longispinosus is a predatory mite (Phytoseiidae) widely used in the biological control of agricultural pests, such as Tetranychus mites, Eriophyes mites and Oligonychus mites, in tropical and subtropical regions in China. It shows strong adaptability to tropical climates and high temperatures. To explore the molecular basis of its excellent properties, such as high-temperature resistance, and to promote its practical application, we conducted a genome sequencing study on N. longispinosus. Here, we report a chromosome-level genome assembly and annotation of the N. longispinosus genome using multiplatform sequencing. The final assembly spanned 199.21 Mb, close to the 210.09 Mb estimated by 21-mer analysis. The chromosome-level assembly achieved a scaffold N50 of 54.56 Mb and a contig N50 of 2.35 Mb. Hi-C scaffolding anchored 98.59% of the assembled sequences to four pseudochromosomes. Genome annotation identified 22.38% repetitive elements and 12,890 predicted genes in the genome. This high-quality genome provides a robust reference for future studies on biological control employing predatory mites.
Similar content being viewed by others


Background & Summary
Neoseiulus longispinosus is an important predatory natural enemy of the family Tetranychidae in agricultural ecosystems1 and is distributed mainly in tropical and subtropical regions of Asia. Predatory mites can prey on pestiferous spider mites, such as Eotetranychus sexmaculatus, Tetranychus cinnabarinus, Oligonychus coffeae, Panonychus citri, Tetranychus neocaledonicus, and Tetranychus truncatus2,3,4,5. They are small and yellow or red in colour. There is strong sexual dimorphism between adult males and females: females have round tails and larger body sizes (1.5 times greater than those of males), whereas males have flat tails. The application of this mite in agricultural production can reduce the use of chemical pesticides and ensure crop quality and safety, highlighting its outstanding application potential. N. longispinosus has been used to control T. urticae in papaya orchards in south Florida, USA3, and Panonychus citri in citrus orchards in Southeast Asia4. This species is one of the most effective predators of spider mites in tropical and subtropical habitats and may become an integral part of biological pest control6. Research on N. longispinosus has thus far focused on laboratory evaluation of its pest-suppression ability, while genomic information for this species remains unavailable. This prohibits the exploration of the basic mechanism of intrinsic population adaptation and the molecular regulation of pest‒predator interactions, as well as the strengthening of its use in the future. Here, we sequenced and analysed the genome of N. longispinosus to better study and utilise predatory mites in controlled applications.
We used a combination of sequencing technologies to integrate the PacBio7, Illumina, RNA-seq, and Hi-C technologies for N. longispinosus. These approaches generated a high-quality chromosome-level genome assembly (Table 1). The final genome assembly spanned 199.21 Mb, 196.39 Mb of which was anchored on four pseudochromosomes, indicating effective chromosome-level scaffolding. The assembly consisted of 54.56 Mb of scaffold N50 and 2.35 Mb of contig N50, with a QV score of 38.69 and a k-mer-based completeness of 90.68%, indicating the high continuity and quality of the assembly. Repetitive elements accounted for 22.38% of the genome. The annotation identified 12,890 protein-coding genes, with an average protein length of 493.2 amino acids. These results provide a foundational genomic resource for N. longispinosus research. Genome information can facilitate the study of the molecular mechanisms of population adaptation and support the future development of large-scale biological control applications.
Methods
Sampling
Neoseiulus longispinosus was collected from soybean leaves in a greenhouse at the China Academy of Tropical Agricultural Sciences, Danzhou, Hainan Province, China. After at least 50 generations of continuous rearing with T. cinnabarinus as prey, a colony of N. longispinosus was used as the source material for genome sequencing. During the sampling process, 2000 eggs were collected and placed into sterile centrifuge tubes, with a total of five tubes being collected. The eggs of N. longispinosus were gently removed from soybean leaves using a fine hairbrush, cleaned of impurities, and subsequently transferred into centrifuge tubes for sequencing.
DNA isolation and sequencing
Genomic DNA was isolated from N. longispinosus eggs using the CTAB method8. DNA concentration was quantified with a Qubit2.0 fluorometer (Thermo Fisher, USA) and a Nanodrop 2000c spectrophotometer (Thermo Fisher, USA) for verification. The quality and integrity of the genomic DNA were assessed by 1% agarose gel electrophoresis. Sample purity was determined using spectrophotometric ratios (260/280 and 260/230). Qualified DNA was fragmented into 15-kb fragments using a Megaruptor instrument (Diagenode B06010001). The resulting fragments were subsequently used for library construction using a SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, Cat. #PN 101-853-100; Menlo Park, CA, USA). Finally, the library was sequenced on a PacBio Sequel II platform. For Illumina sequencing, a 350-bp insert library was constructed utilising a TruSeq DNA PCR-Free Library Preparation Kit (Illumina, Cat. #20015962; San Diego, CA, USA), which was followed by paired-end sequencing on an Illumina NovaSeq. 6000 platform. All library preparation and sequencing operations were performed by Berry Genomics Corporation (Beijing, China).
Genome size estimation
Illumina paired-end reads yielded 11.35 Gb of raw data (Table 2). Quality control and trimming were performed using BBTools (v38.82)9. Adapter contamination was addressed first by trimming with bbduk.sh, followed by the elimination of duplicate reads using “clumpify.sh” from BBTools (v38.82) to ensure high-quality sequencing data. Additionally, bbduk.sh was employed to filter out low-quality bases (Phred score < 20), remove reads shorter than 15 bp, trim homopolymeric tails (A/G/C) exceeding 10 bp in length, and resolve overlaps in paired-end reads through base correction.
Initially, we generated a k-mer frequency distribution from the quality-controlled Illumina reads utilising khist.sh (BBTools) with a k-mer length of 21. Further k-mer analysis was performed using GenomeScope (v2.0)10 with the following specified parameters: -k 21 -p 2 -m 1000. This analysis predicted a genome size of 210.09 Mb (Table 3). It also estimated a heterozygosity rate of 0.78%, which was visualised as a distinct peak to the left of the main coverage peak in the distribution, and repetitive sequences accounted for 20.33 Mb (9.68%) (Fig. 1).

GenomeScope size estimates for Neoseiulus longispinosus.
Genome assembly
High-quality PacBio HiFi reads, generating 17.03 Gb of sequencing data, were first assembled de novo using Hifiasm (v0.16.1)11 to generate an initial set of contigs. To further refine the assembly and remove heterozygous duplications, first aligned the HiFi reads back to the assembled contigs using Minimap2 (v2.4) to generate a SAM file, which was then converted to the BAM format using SAMtools (v1.15)12 for subsequent processing. This alignment file was then processed with Purge_Dups (v1.2.5)13,14 to detect and remove heterozygous duplicated sequences. Then filtered the refined assembly by discarding any contigs with a sequencing depth below 10 × that were most likely contaminated or erroneous.
A Hi-C library was constructed using an EpiTect Hi-C Kit (QIAGEN, Cat. #59971, Hilden, Germany). The target cells were cross-linked with 1% paraformaldehyde at room temperature for 30 min and then quenched with 0.125 mol/L glycine for 5 min. After cell lysis, the cross-linked chromatin was released and digested using HindIII; the digested fragments were subjected to end repair and biotin labelling and then ligated using Hi-C Ligation Buffer for intramolecular ligation. After reverse cross-linking and DNA purification by protease K digestion, the products were mechanically sheared to a main peak of 350 bp, and the biotin-labelled fragments were enriched using streptavidin magnetic beads. Illumina Adapters were added to complete the library construction. After quality control, the library was sequenced on an Illumina NovaSeq platform (PE150), generating 12.44 Gb of raw data. Chromosome-level assembly of the genome was achieved through the integration of Hi-C data. The Hi-C reads were first processed using Juicer (v1.6.2)15. The deduplicated assembly was then scaffolded using 3D-DNA (v180922)16, with default parameters applied to generate the draft assembly.
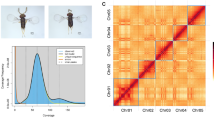
To assess potential contamination in the genome assembly results, a blastn-like search was performed using MMseq. 2 (v13)17 with alignments against the NCBI nt and UniVec databases. Genome completeness was evaluated via BUSCO (v5.4.4)18 using the arachnida_odb10 reference dataset. The draft genome assembly was visualised and manually curated in Juicebox (v1.11.08) to fix the assembly errors. The updated scaffolds were reintroduced into 3D-DNA for final anchoring and ordering to achieve chromosome-level genome assembly (Fig. 2). To assess the quality of the de novo assembly, Merqury (v1.3)19 was used to calculate the QV score and k-mer completeness.

Hi-C intrachromosomal contact map of the Neoseiulus longispinosus genome assembly.
The final genome assembly of N. longispinosus was 199.21 Mb, with a GC content of 50.24%, a QV value of 49.4, and a k-mer completeness of 90.68%. This genome assembly contained 220 contigs, which were divided into 73 scaffolds. The contig N50 was 2.35 Mb, and the scaffold N50 was 54.56 Mb. The longest scaffold length was 60.60 Mb. Overall, 98.59% (196.39 Mb) of the assembly was anchored to four pseudochromosomes (Table 4).
Genome assessment
To evaluate the completeness and base-level accuracy of the assembly, minimap2 was used to realign the PacBio HiSeq sequencing data to the final genome assembly data. SAMtools12 was used to process and analyse the alignment. The results revealed that the average sequencing depth of the whole genome was 85.51 × (Table 5). In addition, the assembly coverage rate of at least one read was 99.69%, indicating a high completion rate. The deep and near-total coverage confirms that the assembly is well supported by the raw sequencing data.
The completeness of the genome assembly was quantitatively evaluated using BUSCO (v5.4.4) with the arachnida_odb10 dataset containing 2,934 conserved single-copy orthologues of Arachnida. BUSCO analysis was performed in genome mode for the assembled genome sequences and protein mode for the predicted protein sequences. The target sequences were aligned against the conserved orthologue set, and the matches were classified into complete (single-copy and duplicated), fragmented, and missing categories on the basis of sequence alignment coverage and homology thresholds.
The completeness of the genome assembly was subsequently evaluated and verified by BUSCO using arachnida_odb10 as the database20. The results revealed that C:96.4% [S: 94.0%, D: 2.4%], F: 0.6%, M: 3.0%, and n: 2934 (Table 6).
To evaluate the integrity of the assembly and confirm the effective use of all the generated original sequencing data, we realigned reads from the three sequencing datasets to the final assembly. To this end, the remapping percentages of Illumina short reads, RNA-seq reads and PacBio HiFi long reads were 94.74%, 92.88%, and 98.78%, respectively (Table 7). The uniformly high mapping rate indicates that most of the raw read data are represented and used in the final assembly, confirming its high degree of integrity.
Therefore, the high BUSCO score, high remapping rates across multiple data types, and chromosome-level continuity (scaffold N50 = 54.56 Mb) confirm that the genome assembly sample has high accuracy and integrity.
Genomic repeat annotation
De novo identification of repetitive elements was initiated using RepeatModeler (v2.0.4) with an additional long terminal repeat (LTR) structure prediction module (-LTRStruct)21, enabling the detection of LTR retrotransposons based on their characteristic structural features. This analysis generated a species-specific repeat database derived from genome sequence homology analysis and ab initio structural motif prediction. To increase the coverage of known repetitive elements, we focused on Arachnida-specific curated repeats that matched the taxonomic identity of N. longispinosus and expanded the de novo library by incorporating sequences from the Dfam 3.522 and RepBase-2018102623 databases, generating a custom repeat library. This custom repeat library was then used for genome-wide repeat masking via RepeatMasker (v4.1.4)24. A total of 245,257 repetitive sequences (44,582,183 bp) were obtained from N. longispinosus, with a repetitive sequence proportion of 22.38%. The five most common types of repeats were unknown (14.27%), DNA transposons (2.95%), LTRs (1.74%), LINEs (1.35%), and simple repeats (0.64%). The genomic structure and annotation were visualised as a circular map with TBTools (v1.098769)25 (Fig. 3).

Circular visualisation of the genomic features of Neoseiulus longispinosus. Note: From outside to inside, chromosome length (Chr), GC content (GC), gene density (GENE), DNA repeat (DNA), short scattered nuclear repeat (SINE), long scattered repeat (LINE), long terminal repeat (LTR) and simple repeat (Simple) are represented.
Prediction of protein-coding genes
Protein-coding gene structure annotation was performed using MAKER (v3.01.03)26, which integrates three types of evidence to increase prediction accuracy. The detailed data were obtained through the following procedure: (1) Ab initio gene prediction: Gene models were predicted utilising BRAKER (v2.1.6)27 and GeMoMa (v1.8)28, incorporating both transcriptomic and proteomic evidence to broaden the pool of potential coding gene candidates, and the prediction results from both tools were subsequently merged to serve as the input file for MAKER ab initio. (2) Transcript alignment-based gene structure prediction: Transcriptomic data were generated by aligning RNA-seq reads to the reference genome using HISAT2 (v2.2.0)29. StringTie (v2.1.6)30 was employed for reference-guided assembly of the second-generation transcriptomic data. (3) Homology-based gene prediction: We downloaded the well-assembled and annotated protein sequences of closely related species on NCBI for homology comparisons: Dermacentor silvarum (GCF_013339745.2)31, Galendromus occidentalis (GCF_000255335.2)32, Tetranychus urticae (GCF_000239435.1)33, Drosophila melanogaster (GCF_00001215.4)34, and Varroa destructor (GCF_002443255.1)35.
MAKER predicted a total of 12,890 protein-coding genes in the N. longispinosus genome, with an average protein length of 493.2 amino acids. The completeness of the predicted protein-coding genes was assessed using BUSCO, with 96.7% complete genes [93.5% single-copy (S) and 3.2% duplicated (D)], 0.2% fragmented (F), and 3.1% missing (M), based on a set of 2,934 conserved orthologues.
Functional annotation of protein-encoding genes
To infer gene functions, protein sequences were compared against the UniProtKB database using the DIAMOND alignment tool (v2.0.11.149)36 with high-sensitivity settings (–very-sensitive -e 1e-5). Functional characterisation was further enhanced by integrating results from multiple comprehensive databases to identify conserved domains, sequence motifs, and biological pathways. Protein domain architecture was analysed using InterProScan (v5.53-87.0)37, which includes key databases, such as Pfam38, SMART39, Superfamily40, and CDD41, to predict structural and functional domains. Additionally, gene functional classification and pathway enrichment were performed using eggNOG-mapper (v2.1.5) against the eggNOG (v5.0) database42,43 for orthology-based annotation.
Finally, 11,478 genes (89.05%) in N. longispinosus matched the UniProtKB database. InterProScan identified 10,190 genes. InterProScan and eggNOG-mapper identified 9,218 GO terms and 4,813 KEGG terms.
Data Records
The raw sequencing reads and genome assembly have been deposited in the NCBI database under BioProject accession number PRJNA124510144. The WGS (SRR35646020), Hi-C (SRR35646023), HiFi (SRR35646022), and RNA-seq (SRR35646021) data are publicly available under accession number SRP62885245. The complete genome assembly is accessible through NCBI with the accession number GCA_052756005.146. Additionally, the genome assembly and annotation files have been made publicly available via Figshare47.
Technical Validation
Genome assembly evaluation
The completeness of the N. longispinosus genome assembly was evaluated using BUSCO against the arachnida_odb10 database. The assembly exhibited exceptional integrity (96.4%), with 94.0% single-copy genes and 2.4% duplicated genes. This finding indicateed that the majority of conserved core genes are present and correctly represented, indicating high gene content integrity.
The continuity of the genome assembly was assessed through multiple metrics. First, the Hi-C heatmap anchoring rate reached 98.59%, indicating that nearly all the assembled sequences were successfully anchored to chromosome-level scaffolds. Second, the scaffold N50 value was 54.56 Mb, reflecting excellent contiguity and a strong ability to assemble long genomic regions into highly continuous scaffolds.
The accuracy of the assembly was confirmed by the high remapping rates of multiple sequencing datasets. Reads from the HiFi, Illumina, and RNA-seq reads exhibited high alignment rates back to the assembled genome, demonstrating strong consistency between the assembly and original sequencing data. This finding supports the reliability and precision of the final genome sequence.
In summary, the N. longispinosus genome assembly demonstrates high levels of completeness, continuity, and accuracy, resulting in a high-quality, chromosome-level genome resource suitable for downstream evolutionary and functional studies.
Data availability
The data presented in this manuscript have not been previously published in any form. The raw sequencing reads and the assembled genome for this project are available in the NCBI database under BioProject accession number PRJNA1245101. The corresponding genome annotation files have been deposited in Figshare (https://doi.org/10.6084/m9.figshare.30164665).
Code availability
No custom code was used in this study. All data analyses were conducted using published bioinformatics software with default settings, unless otherwise specified.
References
Bounfour, M. & Mcmurtry, J. Biology and ecology of Euseius scutalis (Athias-Henriot)(Acarina: Phytoseiidae). J. Hilgardia 55(5), 1–23, https://doi.org/10.3733/hilg.v55n05p023 (1987).
Bhowmik, S. & Yadav, S. K. Neoseiulus longispinosus (Evans)-blessing of Phytoseiids. J. INT J TROP INSECT SC 41, 927–932, https://doi.org/10.1007/s42690-020-00385-4 (2021).
İsmail, D., Alexandra, M., Revynthi, C. K. & Daniel, C. Interactions among exotic and native phytoseiids (Acari: Phytoseiidae) affect biocontrol of two-spotted spider mite on papaya. J. Biological Control. Volume 163, 2021, 104758, ISSN 1049–9644, https://doi.org/10.1016/j.biocontrol.2021.104758 (2021).
Mondal, P., Gowda, C, C. & Srinivasa, N. Comparative biology and demography of the predatory mite Neoseiulus longispinosus (Evans) on five prey species of Tetranychus (Acari: Phytoseiidae, Tetranychidae). J. J Entomol Zool Stud. 8(3), 606–614 (2020).
Huyen, L. et al. Life table parameters and development of Neoseiulus longispinosus (Acari: Phytoseiidae) reared on citrus red mite, Panonychus citri (Acari: Tetranychidae) at different temperatures. J. SYST APPL ACAROLUK. 22(9), 1316–1326, https://doi.org/10.11158/saa22.9.3 (2017).
Jyothis, D. & Ramani, N. Evaluation of prey stage preference of the phytoseiid predator, Neoseiulus longispinosus (Evans) to the spider mite pest, Tetranychus macfarlanei Baker and Pritchard (Acari: Tetranychidae). J. CARYOLOGIA. 59(4), 484–491, https://doi.org/10.11646/zoosymposia.22.1.69 (2023).
Xie, H. et al. PacBio Long Reads Improve Metagenomic Assemblies, Gene Catalogs, and Genome Binning. J. Front Genet. 11, 516269, https://doi.org/10.3389/fgene.2020.516269 (2020).
Gelvin, S. B. & Schilperoort, R. A. Plant molecular biology manual. Springer (2012).
Bushnell, B. BBtools, version 38.82. Retrieved from https://sourceforge.net/projects/bbmap/ (2014).
Ranallo-Benavidez, T. R. et al. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications. 11(1), 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. J. Nat Methods. 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Li, H. et al. The Sequence Alignment/Map Format and SAMtools. J. Bioinformatics. 25(16), 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. J. Bioinformatics. 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. J. Bioinformatics. 34(18), 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-Cexperiments. J. Cell Syst. 3(1), 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. J. Sci. 356(6333), 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Steinegger, M. & Söding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. J. Nat Biotechnol. 35, 1026–1028, https://doi.org/10.1038/nbt.3988 (2017).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. J. Mol Biol Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. J. Genome Biol. 21(245), 9, https://doi.org/10.1186/s13059-020-02134- (2020).
Simao, F. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. J. Bioinformatics. 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Flynn, J. et al. RepeatModeler2 for automated genomic discovery of transposable element families. J. Proc Natl Acad Sci USA. 117(17), 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Hubley, R. et al. The Dfam database of repetitive DNA families. J. Nucleic Acids Res. 44(D1), D81–D89, https://doi.org/10.1093/nar/gkv1272 (2016).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. J. Mobile DNA. 6(11), 1–6, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker, version 4.1.4. Retrieved from http://www.repeatmasker.org (2013–2015).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. J. Mol Plan. 13(8), 1194–1202, https://doi.org/10.1016/j.molp.2020.06.009 (2020).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. J. BMC Bioinform. 12(1), 491, https://doi.org/10.1186/1471-2105-12-491 (2011).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: unsupervised RNA Seq-Based genome annotation with GeneMark-ET and AUGUSTUS. J. Bioinformatics. 32(5), 767–769, https://doi.org/10.1093/bioinformatics/btv661 (2016).
Keilwagen, J. et al. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol Biol. 1962, 161–177, https://doi.org/10.1093/nar/gkw092 (2019).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. J. Nature Methods. 12(4), 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. J. Genome Biol. 20(1), 278, https://doi.org/10.1186/s13059-019-1910-1 (2019).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_013339745.2/ (2022).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000255335.2/ (2012).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000239435.1/ (2011).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001215.4/ (2014).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_002443255.1/ (2017).
Buchfink, B. et al. Sensitive protein alignments at tree-of-life scale using DIAMOND. J. Nature Methods. 18, 366–368, https://doi.org/10.1038/s41592-021-01101-x (2021).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. J. Nucleic Acids Res. 45(D1), D190–D199, https://doi.org/10.1093/nar/gkw1107 (2017).
EI-Gebali, S. et al. The Pfam protein families database in 2019. J. Nucleic Acids Res. 47, D427–D432, https://doi.org/10.1093/nar/gky995 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. J. Nucleic Acids Res. 46(D1), D493–D496, https://doi.org/10.1093/nar/gkx922 (2018).
Wilson, D. et al. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. J. Nucleic Acids Res. 37(Suppl 1), D380–D386, https://doi.org/10.1093/nar/gkn762 (2009).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. J. Nucleic Acids Res. 45, D200–D203, https://doi.org/10.1093/nar/gkw1129 (2017).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. J. Mol Biol Evol. 34(8), 2115–2122, https://doi.org/10.1093/molbev/msx148 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. J. Nucleic Acids Research 47(D1), D309–D314, https://doi.org/10.1093/nar/gky1085 (2019).
NCBI BioProject https://identifiers.org/ncbi/bioproject:PRJNA1245101 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP628852 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_052756005.1 (2025).
Zirui, H. et al. Genome Annotation of Neoseiulus longispinosus. Figshare https://doi.org/10.6084/m9.figshare.30164665 (2025).
Acknowledgements
This research was supported by the following projects: the Ministry of Agriculture and Rural Affairs Free Performance Project (JDZX-2024); the National Natural Rubber Industry Technology System Pest Control Post (CARS-33-BC2); the Central Public-Interest Scientific Institution Basal Research Fund for the Chinese Academy of Tropical Agricultural Sciences (1630042022006,1630042025026); and the Winter Melon and Vegetable Industry Cluster Project of Hainan Province (GJJQ2025DJGC002). Funding Ministry of Agriculture and Rural Affairs Free Performance Project (JDZX-2024); National Natural Rubber Industry Technology System Pest Control Post (CARS-33-BC2); The Central Public-Interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (1630042022006,1630042025026); The Winter Melon and Vegetable Industry Cluster Project of Hainan Province (GJJQ2025DJGC002).
Author information
Authors and Affiliations
Contributions
Jun-Yu Chen provided financial support and technical guidance for this study. Zi-Rui Han and Li-Jiu Zheng. conducted the data analysis, as well as drafted and revised the manuscript. Lang Liang, Zheng-Pei Ye, Yue-Guan Fu, and Tian-Ci Yi revised part of the manuscript text. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, ZR., Zheng, LJ., Liang, L. et al. Chromosome-level genome assembly of Neoseiulus longispinosus (Phytoseiidae). Sci Data 13, 341 (2026). https://doi.org/10.1038/s41597-026-06992-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06992-z
