
Abstract
Nicotiana attenuata and Nicotiana obtusifolia are two wild tobacco species from the Solanaceae plant family, which produce diverse specialized metabolites and have been established as ecological model systems for studying plant-insect interactions. The previously published N. attenuata and N. obtusifolia genomes were draft assemblies with low assembly contiguity and a high proportion of gaps, limiting comparative genomic analysis. Here, we performed chromosome-level genome assemblies and annotations of these two species using long-read sequencing and Hi-C data. Both N. attenuata and N. obtusifolia genomes were anchored to 12 chromosomes, with assembled genome sizes of 2.2 Gb and 1.3 Gb, respectively. We achieved high BUSCO completeness at the protein level, with 99.3% for N. attenuata and 97.9% for N. obtusifolia. The reference genomes of N. attenuata and N. obtusifolia presented in this study will advance the understanding of metabolic innovations in Solanaceae plants.
Similar content being viewed by others
Background & Summary
Nicotiana attenuata and Nicotiana obtusifolia are diploid (2n = 2x = 24) Solanaceae species, which have been developed as ecological model plants to study plant-environment interactions in nature1,2,3. In particular, N. attenuata has been extensively used to understand the function, evolution and diversification of plant specialized metabolites (PSM), such as nicotine, as a defensive response to herbivory and other ecological factors, using genomics, transcriptomics, and metabolomics3,4,5. Further, N. attenuata has been used to understand the balance between plant defence and protection of autotoxicity conferred by the toxic PSMs6. Similarly, N. obtusifolia is also used to study ecological interactions of plants with pathogens, herbivores, and pollinators in natural habitats, along with N. attenuata and others3. Previous omics studies have used N. obtusifolia to understand the genetic basis of metabolic innovation and diversification of plant specialized metabolites in the Nicotiana genus, which are produced as a defence response to environmental stress7,8,9.
With the significance of these species as ecological models, and the necessity of future genomics-guided multi-omics strategies to further understand the metabolic innovations at the level of both plant organ and developmental stage, high-quality genomes are essential for comparative analysis10. A large number of genomic, transcriptomic, and metabolomic datasets already exist for N. attenuata11. However, high-quality genome assemblies were still not available for both of these Nicotiana species. The first published draft genomes of N. attenuata and N. obtusifolia had low assembly contiguities (N50 values of 524.5 Kb and 134.1 Kb, respectively)9. Currently, two genome assemblies of N. attenuata are available, superscaffolded into 12 linkage groups using optical mapping and genetic mapping data (NCBI GenBank accessions: GCF_001879085.1 and GCA_030864195.1); however, several issues remain, including fragmented chromosomes and a high percentage of gaps (Supplementary Table 1).
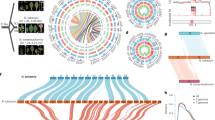
Here, we used the existing N. attenuata genome assembly (GCA_030864195.1, assembled from PacBio reads)12 to reconstruct the chromosomes based on chromatin interaction data obtained from Hi-C sequencing, and report the improved chromosome-level genome assembly of N. attenuata (12 chromosomes, 2.07 Gb) (Fig. 1a, Table 1)13. Compared to the previously available assemblies, our Hi-C data-based assembly showed an increased genomic length covered by the chromosomes, a reduction in the number of ambiguous bases (N), a reduction in the total number and length of gaps, clean Hi-C contact maps with no evidence of mis-joins, and a better accuracy of the genome assembly (Supplementary Table 1). Further, we performed a de novo genome assembly of N. obtusifolia using PacBio sequencing data, and anchored it to chromosomes using Hi-C data, reporting the first chromosome-scale N. obtusifolia genome (12 chromosomes, 1.29 Gb) (Fig. 1b, Table 1). The improvements in the genome assembly statistics compared to the previous contig-level draft assembly of N. obtusifolia are mentioned in Supplementary Table 2.

Circos plots showing the genomic features (window size 100 Kb)13 – (a) N. attenuata, (b) N. obtusifolia. From the outer to inner circle – I. Chromosome length, II. Percentage of repeat regions, III. Percentage of gene regions, IV. GC content.
Repetitive regions contributed to 83.29% and 79.21% of the N. attenuata and N. obtusifolia genome assemblies, respectively. It is noteworthy to mention that the usage of long PacBio reads and the reads obtained from high-throughput chromatin conformation capture (Hi-C) technology enabled the improvement of the assembly quality of such repetitive genomes, which was a limitation in the previous studies9,12. After soft-masking the genome assemblies, we identified 35,166 and 27,352 protein-coding genes in N. attenuata and N. obtusifolia, respectively, using the BRAKER3 pipeline14 (Table 1).
The chromosome-level genome assemblies of N. attenuata and N. obtusifolia presented in this study will serve as a valuable dataset, facilitating a better understanding of the genomic basis of metabolic innovations not only in Nicotiana but also in the Solanaceae family.
Methods
Sample preparation and sequencing
N. attenuata (NCBI TaxID: 49451) plants were grown from Max Planck Institute for Chemical Ecology (MPI-CE) seed stock accession UT31, an inbred ‘UT’ line (31st inbred generation) derived from seeds originally collected at DI Ranch (Santa Clara, Utah, USA) in 1988. UT31 was the same N. attenuata accession that was used in the previous study12. N. obtusifolia (NCBI TaxID: 200316) plants were grown from MPI-CE seed stock Nob02, an inbred line derived from seeds originally collected at Lytle ranch preserve (Santa Clara, Utah, USA) in 2004. The N. attenuata and N. obtusifolia plants used for sequencing were made available as herbarium vouchers at the Johannes Gutenberg University Botanical Garden Herbarium (Herbarium MJG) with accessions MJG 048398 and MJG 048399, respectively (Supplementary Figures 1, 2).
For sequencing, high-quality genomic DNA was extracted from frozen leaf samples using a modified CTAB method15. RNase A was used to remove RNA contaminants. For N. attenuata, a Hi-C fragment library (insert size 300–700 bp) was constructed using the Mate-pair kit after chromatin cross-linking with formaldehyde and enzymatic digestion with DpnII. The library was then sequenced using the Illumina NovaSeq platform. For N. obtusifolia, whole-genome sequencing was performed using the PacBio Revio platform, and for Hi-C data-based contig anchoring, a Hi-C fragment library with 300–700 bp insert size was constructed using the same methods as N. attenuata, and sequenced using the Illumina NovaSeq platform. PacBio Iso-seq RNA-seq library was also constructed using total RNA from the leaf sample, which was sequenced on a PacBio Sequel II platform. The sequencing was carried out at Biomarker Technologies (Beijing, China).
Genome assembly
For N. attenuata, 301.92 Gb clean data were generated from the Hi-C library and were mapped to the previously assembled N. attenuata genome (GCA_030864195.1, assembled from PacBio reads)12 using BWA v0.7.17 with the default parameters16. Invalid read pairs, including Dangling-End and Self-cycle, Re-ligation and Dumped products, were filtered out. LACHESIS was used to cluster, order, and orient the contigs utilising the Hi-C interaction signals17. After Hi-C data-based anchoring, 2.07 Gb (94.6%) of the total assembly (2.19 Gb) was in confirmed order and orientation, constituting the 12 chromosomes. The parameters used in LACHESIS were: CLUSTER_MIN_RE_SITES = 161; CLUSTER_MAX_LINK_DENSITY = 2; ORDER_MIN_N_RES_IN_TRUNK = 199; ORDER_MIN_N_RES_IN_SHREDS = 239.
Prior to N. obtusifolia de novo genome assembly, Illumina paired-end reads from the previous study were quality-filtered using Trimmomatic v0.39 with the parameters “SLIDINGWINDOW:4:15 MINLEN:36”, which were then used to construct the k-mer frequency-based histogram (k-mer = 31) using Jellyfish v2.2.10, and to estimate the genomic characteristics using GenomeScope v2 (Supplementary Figure 3)18,19,20. For the genome assembly, high-accuracy CCS data (96.91 Gb, N50 = 20.67 Kb) were assembled using Hifiasm v0.24.0 with the parameters: l = 0 and n = 4, resulting in 329 contigs (Supplementary Table 3)21. For anchoring of the contigs, 197.74 Gb clean Hi-C data were mapped to the assembly using BWA v0.7.17 with default parameters16. Similar to the N. attenuata genome, the invalid read pairs were filtered out, and the valid interaction read pairs were used for the clustering, ordering, and orienting of scaffolds onto chromosomes with LACHESIS17. After Hi-C data-based anchoring, 1.29 Gb (98.99%) of the total assembly (1.3 Gb) was in confirmed order and orientation, constituting the 12 chromosomes. The parameters used in LACHESIS were: CLUSTER_MIN_RE_SITES = 723; CLUSTER_MAX_LINK_DENSITY = 2; ORDER_MIN_N_RES_IN_TRUNK = 15; ORDER_MIN_N_RES_IN_SHREDS = 15.
The unplaced contigs from both N. attenuata and N. obtusifolia genomes were further analysed using BlobTools to check for contamination22. For this analysis, first, the Illumina paired-end data from the previous study9 were quality-filtered using Trimmomatic v0.39 with the parameters “SLIDINGWINDOW:4:15 MINLEN:36”18. The filtered paired-end reads were mapped onto the respective Nicotiana contigs using BWA-MEM16, and the contigs were mapped against the NCBI-nt database using BLASTN, which were then used to construct the blobplots. Only the contigs that were assigned as “Streptophyta”, “undef”, and “no-hit” were retained (Supplementary Figures 4, 5).
Genome annotation
Both N. attenuata and N. obtusifolia genome assemblies were annotated using a similar method. Prior to coding gene prediction, the genome assemblies were used to construct the corresponding de novo repeat libraries using RepeatModeler v2.0.2, with “-LTRStruct” and other default parameters23. The resultant repeat libraries were used to soft-mask the respective genome assemblies using RepeatMasker v4.1.2 (https://www.repeatmasker.org).
The soft-masked Nicotiana genome assemblies were used for the prediction of high-confidence protein-coding genes using BRAKER314, which implements an integrated transcriptome and proteome evidence-based gene prediction in GeneMark-ETP pipeline24. For transcriptome-based evidence of N. attenuata, the RNA-Seq data (Illumina short reads) from the previous study9 were quality-filtered using Trimmomatic v0.39 with the parameters “SLIDINGWINDOW:4:15 MINLEN:36”, and the filtered data were used in BRAKER314,18. Inside the BRAKER3 pipeline, AUGUSTUS25 was also used for the prediction of coding genes, and TSEBRA26 was used for obtaining a combined output of GeneMark-ETP and AUGUSTUS-based gene predictions.
For transcriptome-based evidence of N. obtusifolia, both Illumina short read RNA-Seq data from a previous study3 and the newly generated PacBio Iso-seq ccs data (292,933 reads) were used in two separate BRAKER3 analyses. First, the Illumina short reads were quality-filtered using Trimmomatic v0.39 with the parameters “SLIDINGWINDOW:4:15 MINLEN:36”, and the filtered data were used in BRAKER314,18. Next, the Iso-seq data were mapped onto the genome using Minimap227, which was then used in a separate BRAKER3 analysis. The results from the two BRAKER3 analyses were merged using TSEBRA26.
In all the BRAKER3 analyses for both the Nicotiana species, the protein sequences from the previous draft assembly of N. attenuata9, along with species belonging to Nicotiana and other Solanaceae genera (only chromosome-level assemblies were considered) were used as a set of extrinsic proteome evidence in GeneMark-ETP. These Solanaceae species were - N. sylvestris28, N. tomentosiformis28, N. tabacum28, N. benthamiana29, Capsicum annuum30, Solanum lycopersicum31, Physalis pruinosa32, Datura inoxia33, Lycianthes biflora33, Lycium chinense34, and Iochroma cyaneum35.
The Nicotiana coding gene sets were filtered to extract the longest isoforms for each gene model and to remove the coding genes with a length of <100 bp using AGAT (https://github.com/NBISweden/AGAT). Further, the coding genes with a repeat content of >50% were filtered out, using a method similar to a previous study36. The protein-coding genes were annotated using the eggNOG-mapper genome annotation server, which uses orthology relationships to assign the KO (KEGG Orthology) terms, GO (Gene Ontology) terms, COG (Cluster of Orthologous Gene) categories, CAZy families, and Pfam domains37. Additionally, the coding genes were also mapped against the UniRef90 database using Diamond v2.0.13 with the parameters: “-k 1 -e 0.00001 --f 6–sensitive”38,39.
Evaluation of genome assembly and annotation
Merqury v1.3 was used to assess assembly completeness and accuracy by comparing k-mers from the Nicotiana genome assemblies and the quality-filtered Illumina paired-end reads, with a k-mer size of 21, as predicted by the “best_k.sh” script9,40. Further, the LTR Assembly Index (LAI) score41 was estimated for the chromosome-level genomes of N. attenuata and N. obtusifolia using GenomeTools42 v1.6.1 and LTR_retriever43 v3.0.1.
To evaluate the completeness of the whole-genome assembly and the predicted coding gene sets, BUSCO v5.4.3 was used in “genome” and “proteins” modes, respectively, with the solanales_odb10 database44. The quality of Nicotiana gene model annotations was assessed using GAQET2 to provide metrics produced by AGAT, OMArk, and PSAURON45,46,47.
Data Records
The newly generated sequencing data for N. attenuata have been deposited in the NCBI database with BioProject accession number PRJNA1245670. The Hi-C genomic reads of N. attenuata are available under SRA accession SRR3296404348. Genome and transcriptome sequencing raw data of N. obtusifolia generated in this study have been deposited in the NCBI database with BioProject accession number PRJNA1332718. The SRA accessions for the N. obtusifolia sequencing reads are - SRR35556615 (PacBio Revio genomic reads)49, SRR35556614 (Hi-C genomic reads)50, and SRR35556613 (PacBio Iso-seq transcriptome reads)51. The Whole Genome Shotgun projects have been deposited at DDBJ/ENA/GenBank under the accession JBUBWS00000000052 and JBUCPO00000000053 for N. attenuata and N. obtusifolia, respectively. The versions described in this paper are versions JBUBWS010000000 and JBUCPO010000000 for N. attenuata and N. obtusifolia, respectively. The genome assembly and annotation files are also available at Figshare54,55. The mapping results of the Nicotiana gene sets constructed in this study against those of the previous study9 are also available at Figshare54,55.
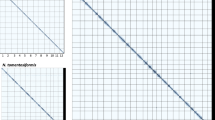
Technical Validation
To evaluate the genome assembly quality and completeness, we implemented multiple strategies. First, the Hi-C contact map showed strong intra-chromosomal interaction signals along the diagonal, which confirms the genome structure integrity (Fig. 2). Second, Merqury analysis results showed the k-mer completeness and QV (consensus quality) values of 96.53% and 42.13, respectively, for N. attenuata, and 98.35% and 48.22, respectively, for N. obtusifolia (Supplementary Figures 6, 7). Third, the chromosome-level genome assemblies of N. attenuata and N. obtusifolia had LAI scores of 14.6 and 15.6, respectively, which refer to “Reference”-standard genome assemblies, according to Ou et al.41 (Fig. 3). Fourth, BUSCO analysis showed the presence of 98.5% complete BUSCO genes in both N. attenuata and N. obtusifolia genome assemblies (Table 2). At the proteome level, the BUSCO completeness were 99.3% and 97.9% for N. attenuata and N. obtusifolia, respectively (Table 2). Fifth, the coding gene sets showed PSAURON quality scores of 96.1 and 96.7 for N. attenuata and N. obtusifolia, respectively. Detailed quality metrics for the genome annotation are provided in Supplementary Table 4.

Hi-C contact maps of the genome assemblies – (a) N. attenuata, (b) N. obtusifolia.

Distribution of the LAI scores in the 12 chromosomes of – (a) N. attenuata, (b) N. obtusifolia.
Data availability
The newly generated sequencing data for N. attenuata have been deposited in the NCBI database with BioProject accession number PRJNA1245670. The Hi-C genomic reads of N. attenuata are available under SRA accession SRR32964043 (https://identifiers.org/ncbi/insdc.sra:SRR32964043). Genome and transcriptome sequencing raw data of N. obtusifolia generated in this study have been deposited in the NCBI database with BioProject accession number PRJNA1332718. The SRA accessions for the N. obtusifolia sequencing reads are - SRR35556615 (PacBio Revio genomic reads) (https://identifiers.org/ncbi/insdc.sra:SRR35556615), SRR35556614 (Hi-C genomic reads) (https://identifiers.org/ncbi/insdc.sra:SRR35556614), and SRR35556613 (PacBio Iso-seq transcriptome reads) (https://identifiers.org/ncbi/insdc.sra:SRR35556613). The Whole Genome Shotgun projects have been deposited at DDBJ/ENA/GenBank under the accession JBUBWS000000000 (https://identifiers.org/ncbi/insdc:JBUBWS000000000) and JBUCPO000000000 (https://identifiers.org/ncbi/insdc:JBUCPO000000000) for N. attenuata and N. obtusifolia, respectively. The versions described in this paper are versions JBUBWS010000000 and JBUCPO010000000 for N. attenuata and N. obtusifolia, respectively. The genome assembly and annotation files are also available at Figshare (https://doi.org/10.6084/m9.figshare.30505763.v3 and https://doi.org/10.6084/m9.figshare.30505793.v2). The mapping results of the Nicotiana gene sets constructed in this study against those of the previous study are also available at Figshare.
Code availability
All bioinformatic analyses were performed according to the manuals provided by the software developers. The software versions and the parameters used for the analyses are mentioned in the “Methods” section. The code used in this study is available on GitHub at https://github.com/Xu-lab-Evolution/Nicotiana_genome_project.
References
Schuman, M. C., Barthel, K. & Baldwin, I. T. Herbivory-induced volatiles function as defenses increasing fitness of the native plant Nicotiana attenuata in nature. eLife 1, e00007 (2012).
Choung, S. et al. MYC2 and MYC3 orchestrate pith lignification to defend Nicotiana attenuata stems against a stem-boring weevil. New Phytol. 247, 2425-2441 (2025).
Zhou, W. et al. Evolution of herbivore-induced early defense signaling was shaped by genome-wide duplications in Nicotiana. eLife 5, e19531 (2016).
Xu, S. et al. Allelic differences of clustered terpene synthases contribute to correlated intraspecific variation of floral and herbivory-induced volatiles in a wild tobacco. New Phytol. 228, 1083–1096 (2020).
Li, D., Heiling, S., Baldwin, I. T. & Gaquerel, E. Illuminating a plant’s tissue-specific metabolic diversity using computational metabolomics and information theory. Proc. Natl. Acad. Sci. USA. 113, E7610–E7618 (2016).
Li, J. et al. Controlled hydroxylations of diterpenoids allow for plant chemical defense without autotoxicity. Science 371, 255–260 (2021).
Elser, D. et al. Evolutionary metabolomics of specialized metabolism diversification in the genus Nicotiana highlights Nacylnornicotine innovations. Sci. Adv. 9, eade8984 (2023).
Kaminski, K. P. et al. Alkaloid chemophenetics and transcriptomics of the Nicotiana genus. Phytochemistry 177, 112424 (2020).
Xu, S. et al. Wild tobacco genomes reveal the evolution of nicotine biosynthesis. Proc. Natl. Acad. Sci. USA. 114, 6133–6138 (2017).
Xu, S. & Gaquerel, E. Evolution of plant specialized metabolites: beyond ecological drivers. Trends Plant Sci. 30, 826-836 (2025).
Brockmöller, T. et al. Nicotiana attenuata Data Hub (NaDH): An integrative platform for exploring genomic, transcriptomic and metabolomic data in wild tobacco. BMC Genomics 18, 79 (2017).
Ray, R. et al. A persistent major mutation in canonical jasmonate signaling is embedded in an herbivory-elicited gene network. Proc. Natl. Acad. Sci. USA. 120, e2308500120 (2023).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. 34, 769–777 (2024).
Abu Almakarem, A. S., Heilman, K. L., Conger, H. L., Shtarkman, Y. M. & Rogers, S. O. Extraction of DNA from plant and fungus tissues in situ. BMC Res. Notes 5, 266 (2012).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv https://arxiv.org/abs/1303.3997 (2013).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114-2120 (2014).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764-770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Challis, R., Richards, E., Rajan, J., Cochrane, G. & Blaxter, M. BlobToolKit – Interactive Quality Assessment of Genome Assemblies. G3 Genes|Genomes|Genetics 10, 1361–1374 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 117, 9451-9457 (2020).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP significantly improves the accuracy of automatic annotation of large eukaryotic genomes. Genome Res. 34, 757–768 (2024).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics 22, 566 (2021).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094-3100 (2018).
Sierro, N., Auberson, M., Dulize, R. & Ivanov, N. V. Chromosome-level genome assemblies of Nicotiana tabacum, Nicotiana sylvestris, and Nicotiana tomentosiformis. Sci. Data 11, 135 (2024).
Ko, S. R. et al. High-quality chromosome-level genome assembly of Nicotiana benthamiana. Sci. Data 11, 386 (2024).
Liu, F. et al. Genomes of cultivated and wild Capsicum species provide insights into pepper domestication and population differentiation. Nat. Commun. 14, 5487 (2023).
Su, X. et al. A high-continuity and annotated tomato reference genome. BMC Genomics 22, 898 (2021).
He, J. et al. Establishing Physalis as a Solanaceae model system enables genetic reevaluation of the inflated calyx syndrome. Plant Cell 35, 351–368 (2023).
Wu, Y. et al. Phylogenomic discovery of deleterious mutations facilitates hybrid potato breeding. Cell 186, 2313–2328.e15 (2023).
Yang, J. et al. Multiple independent losses of the biosynthetic pathway for two tropane alkaloids in the Solanaceae family. Nat. Commun. 14, 8457 (2023).
Powell, A. F. et al. Genome sequence for the blue-flowered Andean shrub Iochroma cyaneum reveals extensive discordance across the berry clade of Solanaceae. Plant Genome 15, e20223 (2022).
Wang, Y. & Xu, S. A high-quality genome assembly of the waterlily aphid Rhopalosiphum nymphaeae. Sci. Data 11, 194 (2024).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115-2122 (2017).
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B. & Wu, C. H. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926-932 (2015).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Gremme, G., Steinbiss, S. & Kurtz, S. Genome tools: A comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinforma. 10, 645-656 (2013).
Ou, S. & Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410-1422 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Garcia-Carpintero, V., de Martín, I., García-Juan, S. & Bombarely, A. Structural Genome Annotation QC with GAQET2. https://doi.org/10.17504/protocols.io.8epv5k5rnv1b/v1 (2025).
Sommer, M. J., Zimin, A. V. & Salzberg, S. L. PSAURON: a tool for assessing protein annotation across a broad range of species. NAR Genomics and Bioinformatics 7, lqae189 (2025).
Nevers, Y. et al. Quality assessment of gene repertoire annotations with OMArk. Nat. Biotechnol. 43, 124–133 (2025).
NCBI sequence read archive https://identifiers.org/ncbi/insdc.sra:SRR32964043 (2025).
NCBI sequence read archive https://identifiers.org/ncbi/insdc.sra:SRR35556615 (2025).
NCBI sequence read archive https://identifiers.org/ncbi/insdc.sra:SRR35556614 (2025).
NCBI sequence read archive https://identifiers.org/ncbi/insdc.sra:SRR35556613 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBUBWS000000000 (2026).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBUCPO000000000 (2026).
Chakraborty, A. & Xu, S. Chromosome-level genome assembly and annotation of Nicotiana attenuata. figshare. Dataset. https://doi.org/10.6084/m9.figshare.30505763.v3 (2025).
Chakraborty, A. & Xu, S. Chromosome-level genome assembly and annotation of Nicotiana obtusifolia. figshare. Dataset. https://doi.org/10.6084/m9.figshare.30505793.v2 (2025).
Acknowledgements
We thank Dr. Danny Kessler from the Max Planck Institute for Chemical Ecology for providing the plant materials. We thank Dr. Patrycja Baraniecka and Thoomke Roth for preparing the plant materials for sequencing. We are grateful for the help of Dr. Christian Siadjeu, Dr. Ralf Omlor, and Markus Hageneuer in preparing the voucher specimens of N. attenuata and N. obtusifolia. Parts of this research were conducted using the supercomputer Mogon 2 and/or advisory services offered by Johannes Gutenberg University Mainz (hpc.uni-mainz.de), which is a member of the AHRP (Alliance for High-Performance Computing in Rhineland Palatinate, www.ahrp.info) and the Gauss Alliance eV. The authors gratefully acknowledge the computing time granted on the supercomputer MOGON 2 at Johannes Gutenberg University Mainz (hpc.uni-mainz.de). This work was supported by the Agence Nationale de la Recherche (ANR)–Deutsche Forschungsgemeinschaft (DFG) joint research funding for French–German Collaboration (ANR-23-CE20-0037 to E.G. and DFG project number 529944545 to S.X.).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.C. conducted the research and performed the data analysis. S.X. conceived and supervised the study. Both authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakraborty, A., Xu, S. Chromosome-level genome assemblies of Nicotiana attenuata (coyote tobacco) and Nicotiana obtusifolia (desert tobacco). Sci Data 13, 441 (2026). https://doi.org/10.1038/s41597-026-07080-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-07080-y
