
Abstract
The COVID-19 pandemic has been marked by novel viral variants, posing challenges to global public health. Recombination, a viral evolution mechanism, is implicated in SARS-CoV-2's ongoing evolution. The XBB recombinant lineage, known for evading antibody-mediated immunity, exhibits higher transmissibility without increased disease severity. We investigated the prevalence and genomic features of XBB in SARS-CoV-2-positive cases in Rio Grande do Sul (RS), Brazil. We sequenced 357 samples from epidemiological weeks (EW) 47/2022 to 17/2023, and included 389 publicly available sequences. Clinical and epidemiological data were obtained from DATASUS, e-SUS, and SIVEP GRIPE (data recording systems of the Brazilian Ministry of Health). Of these, 143 were classified as XBB and 586 were other Omicron lineages. In March 2023 (EW 10), XBB became dominant, accounting for 83.3% of cases. 97.7% of XBB-infected patients successfully recovered from the infection, with a low mortality rate (2.3%). Even after receiving three vaccine doses and having been previously infected, 59.5% of the patients experienced reinfection with XBB. However, for 54% of the individuals, the interval between their XBB infection and the last vaccine dose exceeded one year, potentially leading to a decline in antibody levels. In addition, we identified 90 mutations in RS circulating XBB, spread throughout the genome, notably in the Spike protein region associated with immune resistance. This study provides insights into the dynamics and impact of a recombinant variant becoming predominant for the first time in the state. Continued surveillance of SARS-CoV-2 genomic evolution is crucial for effective public health management.
Similar content being viewed by others


Introduction
COVID-19 pandemic has been marked by the continuous evolution of SARS-CoV-2, with the emergence of novel viral variants that pose a threat to global public health. Recombination cannot be overlooked as it is a main contributor to viral evolution1. In SARS-CoV-2, recombination events require co-circulation and co-infection of different lineages in the same host. Within the cell infected with genetically different lineages, it is hypothesized that viral RNA-dependent RNA polymerase (RdRp) detaches from a template with the nascent RNA strand intact and resumes elongation in another template2. XBB is a recombinant lineage of BJ.1 (also known as BA.2.10.1) and BM.1.1.1 (also known as BA.2.75), both belonging to the BA.2 lineage. XBB and XBB.1 were first identified in India in August 2022, have spread rapidly in several Asian countries and are rising worldwide1. The XBB lineage, along with its descendants XBB.1, XBB.1.5, and XBB.1.16, are playing a role in the recent surges observed in different nations. As of October 12, 2022, XBB* was classified as Variant Under Monitoring (VUM). XBB.1.5 and XBB.1.16 are Variants of Interest (VOI) since January 11th and April 17th, 2023, respectively3.
XBB and its descendant, XBB.1, have the capacity to evade antibody-mediated immunity conferred by vaccination or previous infection4. This lineage carries multiple mutations, including those in the Spike protein, which are responsible for its ability to evade the immune response, and resistance to several monoclonal antibodies classes5. In fact, XBB recombination breakpoint is located within the Receptor-Binding Domain (RBD) of the S gene. Multiple mutations in the RBD and the N-terminal domain (NTD) are associated with XBB immune resistance6. Fully vaccinated individuals remain protected from hospitalization and death4.
In Brazil, XBB was first detected in October 2022. Initially, XBB* were a minor component of the viral landscape, making up less than 10% of the circulating lineages7. However, the XBB* lineages, particularly those with the F486P mutation, rapidly gained prevalence from January to February 2023, becoming the dominant lineage in the country. The surge was characterized by the stepwise accumulation of mutations in the spike protein's receptor-binding domain (RBD), including F486P, F456L, and L455F, which enhanced the virus's ability to evade immunity and bind to the ACE2 receptor. In November 2022, the XBB* + F486P group was first detected. This was followed by the emergence of the XBB* + F486P + F456L group in January 2023, and subsequently, the XBB* + F486P + F456L + L455F group in March 2023. These groups sequentially replaced their predecessors, each demonstrating a higher relative reproduction rate, indicating a notable evolutionary advantage7.
The emergence of a new lineage raises concerns regarding its immunoevasion properties5. While XBB and its sublineages do not cause more severe disease compared to other Omicron lineages, they exhibit higher transmissibility, leading to increased rates of infections and reinfections. Consequently, this results in a rise in the number of confirmed cases, hospitalizations, and deaths8,9. Due to its genomic characteristics, prevalence, immune evasion potential, and mutations affecting infection, continuous surveillance and monitoring are essential.
Here, we investigate the prevalence and genomic characteristics of the first recombinant variant that predominated in SARS-CoV-2-positive cases in the RS state, Brazil. We compiled 357 samples positive for SARS-CoV-2 that were sequenced, as well as 389 publicly available sequences collected from November 20, 2022 to April 29, 2023. Our study sheds light on the dynamics and potential impact of recombinant variants on the COVID-19 and highlights the urgent need for continued monitoring SARS-CoV-2 genomic evolution.
Materials and methods
Study design
This is a cross-sectional study conducted with the approval of the Research Ethics Committee of the Universidade Federal de Santa Maria (CAAE: 51019821.0.0000.5346), ensuring compliance with ethical guidelines.
Participants
Our laboratory composed two subprojects of the "Rede Vírus”, an initiative by the Brazilian Ministry of Science, Technology, and Innovation. This initiative is centered on the collection and genomic sequencing of SARS-CoV-2 samples, functioning as an integral component of the Rio Grande do Sul (RS) state government's genomic surveillance network. Therefore, this study included SARS-CoV-2-positive samples from patients residing in the RS state, South Brazil, from November 20, 2022, to April 29, 2023, which corresponded to epidemiological weeks (EW) 47/2022 to 17/2023. SARS-CoV-2 infection was confirmed through RT-qPCR tests conducted in both public and private laboratories within the RS state. It is noteworthy that the Brazilian Ministry of Health has issued a Guideline Note in October, 2021 advocating for the utilization of a rapid antigen test to detect SARS-CoV-2 in suspected infection cases. This test is deemed to be more cost-effective and less intricate than RT-qPCR, although it exhibits lower sensitivity10. As a result, the amount of sequenced samples that can be used in genomic surveillance is decreasing.
We also conducted a search for SARS-CoV-2 sequences from RS state deposited in GISAID, focusing on samples collected during the investigation period. In order to ensure data quality, we selected sequences with complete genomes and excluded those with low coverage. The download of sequences was performed on May 19, 2023.
Data sources
Epidemiological and clinical data on the XBB-infected patients were obtained from DATASUS, e-SUS, and SIVEP GRIPE (data recording systems of the Brazilian Ministry of Health) in accordance with patient data protection laws. Our evaluation included data on age; sex; date of the SARS-CoV-2 positive test through RT-qPCR; first infection, reinfection or if there was no information in the databases; patient required hospitalization in the ward or ICU; patient's clinical outcome, i.e., recovered or died; death related to COVID-19 complications or other causes. Additionally, we collected information on vaccination status, including vaccination strategy and schedule.
Sample screening
SARS-CoV-2 positive samples were collected in RS state and sent to Laboratório de Biologia Molecular e Bioinformática Aplicada à Microbiologia Clínica (LABIOMIC), Universidade Federal de Santa Maria, Santa Maria city (n = 357).
Genome sequencing
Sequencing was performed on all samples with a cycle threshold (Ct) ≤ 30, using the ARTIC protocol version 3 (https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bp2l6n26rgqe/v3?version_warning=no, accessed on may 19, 2023), with V4 or V4.1 primer scheme or Illumina COVIDSeq Test (Illumina, San Diego, CA), with V3 primer scheme. Genomes were sequenced using either the MinION device (Oxford Nanopore Technologies, Oxford, UK) or the Illumina iSeq 100. Raw data from the MinION was collected using MinKNOW v.20.10.3 and high-accuracy base-calling and quality control analyses were performed using Guppy v.4.2.2. Illumina raw data was processed on Basespace (https://basespace.illumina.com).
Viral genome assembly
To assemble the consensus sequence on MinION data we used the nCoV-2019 novel coronavirus bioinformatics protocol (https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html, accessed on May 19, 2023) and utilized Minimap2 v.2.1711 and BCFtools. The app DRAGEN COVID Lineage was used to obtain the consensus genome sequence from Illumina data.
To ensure the quality of the data, we excluded sequences that exhibited less than 75% genome coverage breadth and contained more than 3000 N (undetermined nucleotides).
Lineage identification
To analyze the clade, identify the number of gap regions, and determine the lineages of the viral consensus sequences, we employed Nextclade version 2.14.1 (https://clades.nextstrain.org/, accessed on May 22, 2023)12.
Single nucleotide polymorphism identification
To identify single nucleotide polymorphisms (SNPs), insertions, and deletions, we used Geneious Prime™13. In brief, consensus sequences were imported and aligned to the SARS-CoV-2 reference genome (NC_045512.2). The "Find Variations/SNPs" tool was configured to identify variants with a minimum frequency of 0.25.
Statistical analysis
Results were expressed as absolute numbers, percentages, or median ± interquartile range, as appropriate for each analysis. All statistical analyses were performed utilizing Graphpad Prism version 6 (www.graphpad.com). Data regarding the vaccination schedule, clinical outcome and age were plotted on a Sankey chart (https://www.sankeymatic.com/). In this chart the study population was stratified based on the vaccination scheme recommended by the Ministry of Health, yielding four distinct age groups for the subsequent analyses14. Data derived from the interval between last vaccine dose and SARS-CoV-2 infection was analyzed by One-way ANOVA followed by Tukey post-hoc test. The results were considered significant when p ≤ 0.05.
Ethics approval
This study was performed in compliance with the principles of the Declaration of Helsinki. Approval was granted by the Institutional Ethical Review Board of the Universidade Federal de Santa Maria (approval number: 51019821.0.0000.5346).
Consent to participate
The requirement for participants' consent was waived by the Institutional Ethical Review Board of the Universidade Federal de Santa Maria (approval number: 51019821.0.0000.5346).
Results
Our study investigated the prevalence of the SARS-CoV-2 recombinant lineage XBB in the RS state, South Brazil, from November 2022 to April 2023. During this period, the RS state government recorded 265,371 new confirmed cases of SARS-CoV-2 infection and 834 deaths (Fig. 1). We observed a ~ 2.5-fold increase in SARS-CoV-2-positive cases and a 4.8-fold increase in deaths following the introduction of XBB into the state on EW 48/2022. Subsequently, from EW 6/2023 onwards, when the first cases of infection by XBB.1.5 were identified, we observed another increase in these parameters, although with less intensity.

Comprehensive overview of the COVID-19 situation in the Rio Grande do Sul state, Brazil, during epidemiological weeks 40/2022 to 17/2023, as indicated by the number of SARS-CoV-2-positive cases and deaths. The data were obtained from the official website of the Rio Grande do Sul state government, available at https://ti.saude.rs.gov.br/covid19/.
A total of 357 SARS-CoV-2-positive samples were sequenced for this study. However, 17 sequences were excluded due to not meeting the quality criteria (i.e., had less than 75% genome coverage breadth, and/or more than 3000 N, and/or possessing a bad score). Additionally, we incorporated 389 publicly available sequences from the GISAID database (Table S1). A total of 729 SARS-CoV-2 genomes were analyzed to determine the predominant variant during the study period. All of them were identified as the Omicron variant. Among these genomes, 586 sequences were identified as non-recombinant lineages, while the remaining 143 were classified as the recombinant variant XBB*. Complete epidemiological data were available for 99.04% (722) of individuals infected with SARS-CoV-2 (Table 1 and S1).
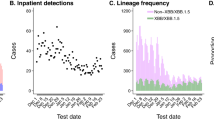
The first case of XBB.1 infection was detected on November 27, 2022 (EW 48/2022) (Fig. 2 and Table S1). Subsequently, we identified a two cases of XBB.1 infection. However, starting from EW 9/2023, the number of cases experienced a substantial increase, ultimately establishing XBB as the predominant lineage, accounting for 83.3% of all cases in EW 10/2023 (Fig. 2A,C, and Table S2). Among 729 SARS-CoV-2 sequences (Fig. 2B,C; Table S1), we identified a total of forty-five Pangolin lineages. The most frequently detected lineage was BQ.1.1 (257 sequences, 35.4%), followed by BE.9 (117 sequences, 16.12%), XBB.1.5 (97 sequences, 13.36%), BQ.1.22 (59 sequences, 8.13%), XBB.1 (32 sequences, 4.41%), BQ.1 (25 sequences, 3.44% each), and BQ.1.1.18 (19 sequences, 2.62%). The remaining thirty-eight lineages had frequencies of less than 2%, collectively representing 16.94% of the total sequences.

SARS-CoV-2 genomic surveillance in the Rio Grande do Sul state, Brazil, from November 2022 to April 2023. (A) Predominance of the XBB over other non-recombinant Omicron lineages, based on the sequenced samples analyzed in this study. Absolute (B) and relative (C) numbers of non- and XBB lineages identified in the samples, categorized by epidemiological weeks, as classified by the Nextclade.
Clinical data was available for 62.2% (89 individuals) of the 143 patients infected with the XBB lineage (Fig. 3A). Among these patients, 97.7% (87 individuals) successfully recovered from the SARS-CoV-2 infection, while 2.3% (two individuals) died due to COVID-19 complications. Only one of the recovered patients required hospitalization (ICU), due to causes other than COVID-19, whereas hospitalization was required for those who died (Table S2). Among the recovered group, 41.6% and 33.7% were aged between 40 and 59 years or > 60 years, respectively. The remaining 24.7% represented individuals up to 11 years old (3 individuals) and those aged 12–39 years (17 individuals). Both individuals who died were over 80 years old, with one having received only two vaccine doses. Notably, five individuals were unvaccinated against SARS-CoV-2, predominantly those in the 0–11 age group, who were not yet eligible for vaccination according to the Ministry of Health guidelines. This group also included one individual aged 12–39 and one over 60, both of whom were eligible but unvaccinated. 41.6% (37 individuals) received three vaccine doses, followed by 27% (24 individuals) who received four doses, and 16.9% (15 individuals) who received two doses. Indeed, only six individuals were up-to-date with their vaccination schedule. The first three doses administered were monovalent vaccines against SARS-CoV-2. Since February 2023, the bivalent vaccine has been introduced, but only 6.7% of individuals have received it.

Clinical outcome and vaccination information for XBB-infected patients. (A) Clinical outcome associated with age and the number of doses and boosters of the vaccine against SARS-CoV-2. (B) Number of infections associated with age and interval between last vaccine and XBB infection.
According to the health data repositories of the Brazilian Ministry of Health, 59.5% of the individuals (50 individuals) were reinfected with the XBB lineage (Fig. 3B). Out of these cases, 4 individuals were reinfected with XBB, 6 with XBB.1, and the majority, 40 individuals, were reinfected with XBB.1.5. A total of 28.6% (24 individuals) experienced their first SARS-CoV-2 infection with this lineage. For 11.9% (10 individuals), there is an absence of recorded data on both previous infections and the XBB infection itself, suggesting a potential failure in the reporting system. 43.1% of the reinfected individuals were over 60 years of age, followed by those in the 40–59 age group (41.2%), and 15.7% aged between 12 and 39 years. Conversely, 50% of those experiencing their first infection were aged between 40 and 59 years. There was considerable variability in the interval between infection and the administration of the last vaccine dose. However, it is noteworthy that 58.3% of the infected individuals had received their last dose or booster more than a year prior. Among the reinfected individuals, all had previously received at least one vaccine dose, indicating the presence of hybrid immunity against the virus.
We analyzed the interval between the occurrence of XBB infection and the administration of the last vaccine dose (Fig. 4). The results revealed that among individuals aged 12–39, 40–60, and > 60 years, the median intervals were 403, 442, and 351 days, respectively. The three infected individuals in the 0–11 age group had not received any vaccine doses.

Clinical outcome and vaccination information for XBB-infected patients according to age. Data were expressed as median ± interquartile interval (n = 17–37) and analyzed by one-way ANOVA followed by Tukey post-hoc test. There were no statistical differences (p > 0.05).
Ultimately, we conducted a SNP analysis to determine if the XBB lineages circulating in RS had acquired any additional mutations and to verify if they exhibited all the characteristic mutations (Fig. 5). The XBB recombinant lineage exhibited a total of 90 mutations, including 78 SNPs, comprising 52 transitions and 26 transversions, 5 substitutions, and 7 deletions (Fig. 5A,B; Table S3). Of these mutations, (1) was located in the 5'-UTR, (31) in the ORF1ab, (40) in the S gene, (3) in ORF3a, (2) in the E gene, (3) in the M gene, (2) in ORF6, (1) in ORF7b, (1) in ORF8, (4) in the N gene, (1) in the intergenic UTR, and (1) in the 3'-UTR (Fig. 4 and Table S3). Among the observed SNPs, 77.2% (61) were non-synonymous mutations, particularly those occurring in the S gene. This accumulation of mutations in the XBB variant's S gene, particularly within the Receptor Binding Domain (RBD), endows it with the ability to evade antibodies acquired through vaccination and previous infections (Fig. 5B).

Mutational profile and variant frequency of the SARS-CoV-2 recombinant variant XBB in the genome coding region (A). The Receptor-Binding Domain (RBD) of the S gene, highlighted by the gray square, encompasses 22 mutations (B). The frequency of each mutation is represented by the height of the corresponding bar.
Discussion
In this study, we conducted an analysis of the initial emergence of a recombinant lineage as the predominant cause of SARS-CoV-2 infections in the RS state. The introduction of the XBB lineage resulted in a substantial ~ 2.5-fold increase in the number of confirmed cases and a 4.8-fold increase in COVID-19 related fatalities. The scientific community has drawn attention to the emergence of recombinant lineages within SARS-CoV-215. While previous circulating recombinant lineages (e.g., XD, XE, XAG, etc.) were detected in a small number of individuals15, XBB and its sublineages have now emerged as predominant. In Brazil, XBB was initially detected in October 20227 and was detected in the RS state at the end of November. To the best of our knowledge, only the study conducted by Arantes et al.7 has assessed the spatiotemporal dynamics and epidemiological impact of XBB in Brazil. The spread of XBB* lineages in the Southern region of Brazil followed a similar temporal pattern to the national trend but exhibited variations in the prevalence of specific XBB* groups. Notably, the dominance of the XBB* + F486P + F456L + L455F group in the Southern region emerged later than in other Brazilian regions.
Vaccinating individuals against SARS-CoV-2 is pivotal in mitigating severe manifestations of COVID-19. A booster dose of Pfizer-BioNTech BNT162b2 has proven efficacious in this regard16,17. However, the advent of the Omicron BA.4 and BA.5 lineages has resulted in a roughly 50% reduction in the vaccine's efficacy against symptomatic disease presentations18. Booster dose's neutralizing capacity is 66 times less effective against the XBB.1.5 strain compared to the WA.1 strain. Additionally, it has been observed that six months post-vaccination, individuals fail to exhibit detectable levels of antibodies against the virus19. Most individuals infected with XBB (78.4%) had only one booster dose given more than a year ago, probably already experiencing a decline, or absence of antibodies. Furthermore, heterologous mRNA vaccine boosters seem to induce a stronger and longer-lasting antibody response against the XBB variant. This highlights the need to understand the mechanisms underlying the differential antibody responses elicited by different vaccine compositions20.
Immunity against SARS-CoV-2 can be induced through vaccination, acquired naturally via infection, or in a hybrid form, resulting from a combination of vaccination and prior infection, has been demonstrated to reduce the risk of reinfection21. In this study, we observed a high reinfection rate of 59.5% among XBB patients. Individuals may suffer reinfection events from SARS-CoV-2 due to several factors, including (i) the increasing number of individuals experiencing a primary SARS-CoV-2 infection; (ii) Some individuals may not develop a strong or lasting immune response22 or over time, immunity acquired from prior infection or vaccination can decline, making some individuals more susceptible to reinfection23; (iii) New variants of SARS-CoV-2, such as Omicron and its lineages, can evade immunity developed from previous infections or vaccinations due to mutations in the spike protein and other regions24. Here, the majority of individuals who experienced reinfection with the new recombinant variant had previously been vaccinated, thus possessing hybrid immunity against SARS-CoV-2, but an extended period had elapsed since vaccination or prior infection. With the data obtained in this study, it is not possible to determine the exact cause of reinfection events. As mentioned, reinfections may result from a complex interaction between immunity, viral evolution, and individual health conditions. This high reinfection rate highlights the need for continued monitoring of variant-specific immune evasion and the effectiveness of existing vaccines. Furthermore, they suggest that future vaccination strategies should consider the development of vaccines targeting specific variants and potentially enhancing booster schemes to maintain robust immunity. Understanding the underlying mechanisms of reinfection can inform public health policies and guide the design of more effective vaccination programs.
The XBB lineage exhibited the accumulation of mutations in the Spike protein, including the BA.2 variant mutations along with an additional fourteen mutations. Among these, five mutations were identified in the NTD, and nine mutations were found in the RBD. Notably, the XBB.1 sublineage demonstrates a specific mutation in this protein, namely G252V. These mutations raise concerns due to their potential to undermine the efficacy of vaccines and monoclonal antibody (mAb) therapies, which rely on targeting specific regions of the Spike protein for neutralization9. Here, we identified a total of 40 mutations in the S gene, with 25% (n = 10) of these mutations being specific to XBB. Among these XBB-specific mutations, five have been previously reported to be linked with immune evasion, namely del14425, R346T26,27, V445P28, F486P29,30, and F490S31,32.
The ORF8 is an accessory protein (121 amino acids) which plays a role in various viral activities, including replication, pathogenesis, and immune evasion. Studies have shown that ORF8 downregulates MHC class I molecules, mimicking interleukin 17-A (IL-17A), and inducing endoplasmic reticulum stress. These mechanisms may explain the cytokine storm associated with the severity of SARS-CoV-2 infection. Throughout the course of the pandemic, mutations in ORF8 have been rapidly evolving and observed in the emergence of new lineages. Deletions in specific parts or the entire ORF8 have been linked to milder disease. Functional and non-mutated ORF8 is associated with higher virulence33,34,35,36. The XBB recombinant variant has the G8* mutation, which incorporates a stop codon closely related to the 5' portion of ORF8. This mutation leads to the premature interruption of the transcription and, consequently, to the translation of a truncated protein. In this case, ORF8-mediated IL-17A mimicry, endoplasmic reticulum stress induction, and MHC class I response suppression will not occur. This XBB-specific mutation (Fig. 5) could be associated with mild to moderate disease cases33,34,35,36. Unfortunately, we did not have access to the patients' clinical data to perform this assessment, which is a limitation of our study.
Although the World Health Organization (WHO) has removed the pandemic status for COVID-19 on May 5, 2023, genomic surveillance plays a pivotal role in tracking viral evolution and promptly identifying new variants, which is crucial for the development of targeted interventions and therapies37. However, several countries, including Brazil, encounter challenges in establishing robust genomic surveillance systems due to limited funding, decentralized coordination, and inadequate data sharing practices38. Therefore, it is imperative to prioritize genomic surveillance as a cornerstone of pandemic preparedness, response, and infectious disease control. Corroborating this, the present study is subject to limitations. Due to the lack of coordinated and standardized sampling criteria from the health system and government, we used convenience sampling. The sequenced samples were those that tested positive by RT-qPCR and were subsequently sent to our laboratory for sequencing. Furthermore, in October 2021, the Brazilian Ministry of Health issued a Guideline Note recommending rapid antigen tests for suspected SARS-CoV-2 cases. This change led to a substantial decrease in the number of samples available for genomic sequencing, resulting, for example, in a data gap during SE 7/2023. Also, our study is geographically limited to evaluating the state of RS, located in the extreme south of Brazil. Despite these limitations, we believe our findings contribute to understanding the pandemic's trajectory in Brazil and offer valuable insights for genomic surveillance.
To combat SARS-CoV-2 effectively, public health strategies must encourage booster doses and prioritize these for high-risk groups such as the elderly and those with underlying health conditions. Improving the reporting system for accurate and timely recording of cases and reinfections is crucial for better public health responses. Increasing funding for genomic surveillance will ensure continuous monitoring of variants. Establishing a centralized genomic surveillance system and integrating different data systems (e.g., DATASUS, e-SUS, SIVEP GRIPE) will streamline data collection and facilitate real-time access to epidemiological and clinical data.
Conclusion
The findings of our study provide insights into the first time that a recombinant lineage predominated in SARS-CoV-2 positive cases in the RS state, Brazil. Introduction of the XBB lineage coincided with an increase in the number of confirmed COVID-19 cases, implying a potential association between the new lineage and increased transmissibility. Clinical outcomes among XBB-infected patients further emphasized the importance of vaccination. While the vast majority of XBB-infected patients recovered, with none requiring hospitalization, most of these patients had received their last vaccine dose or booster more than a year prior, suggesting a possible waning of vaccine-induced immunity over time. Mutation profile of the XBB lineage needs monitoring, given its potential to influence viral transmission, immune evasion, and pathogenicity. These mutations, particularly within the S gene, may impact the effectiveness of current therapeutic interventions and vaccines against SARS-CoV-2. The detection and characterization of the XBB lineage highlight the necessity for rigorous, ongoing genomic surveillance and research. Maintaining an updated vaccination schedule and utilizing genomic surveillance are instrumental in controlling the spread and impact of emerging SARS-CoV-2 variants.
Data availability
The datasets generated and/or analyzed during the current study are available on https://gisaid.org/ (EPI_SET_240315sp), and the access codes for the sequences are provided in Table S1. COVID-19 cases and deaths were obtained from https://ti.saude.rs.gov.br/covid19/. Epidemiological and clinical data were obtained from DATASUS, e-SUS, and SIVEP GRIPE (data recording systems of the Brazilian Ministry of Health), in accordance with patient data protection laws, and can be consulted in Tables S1 and S2. The datasets generated and/or analyzed during the current study are available on https://gisaid.org/ (EPI_SET_240315sp), and the access codes for the sequences are provided in Table S1. COVID-19 cases and deaths were obtained from https://ti.saude.rs.gov.br/covid19/. Epidemiological and clinical data were obtained from DATASUS, e-SUS, and SIVEP GRIPE (data recording systems of the Brazilian Ministry of Health), in accordance with patient data protection laws, and can be consulted in Tables S1 and S2.
References
Scarpa, F. et al. Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages. J. Med. Virol. 95(3), e28625. https://doi.org/10.1002/jmv.28625 (2023).
Focosi, D. & Maggi, F. Recombination in coronaviruses, with a focus on SARS-CoV-2. Viruses 14(6), 1239. https://doi.org/10.3390/v14061239 (2022).
Meyer, S. et al. Prevalent and immunodominant CD8 T cell epitopes are conserved in SARS-CoV-2 variants. Cell Rep 42(1), 111995. https://doi.org/10.1016/j.celrep.2023.111995 (2023).
Imai, M. et al. Efficacy of antiviral agents against omicron subvariants BQ.1.1 and XBB. N. Engl. J. Med. 388(1), 89–91. https://doi.org/10.1056/NEJMc2214302 (2023).
Tamura, T. et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 14(1), 2800. https://doi.org/10.1038/s41467-023-38435-3 (2023).
Arantes, I. et al. Spatiotemporal dynamics and epidemiological impact of SARS-CoV-2 XBB lineage dissemination in Brazil in 2023. Microbiol. Spectr. 12(3), e0383123. https://doi.org/10.1128/spectrum.03831-23 (2024).
Vogel, L. What to know about Omicron XBB.1.5. CMAJ 195(3), 127–128. https://doi.org/10.1503/cmaj.1096034 (2023).
Wang, Q. et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 186(2), 279–286. https://doi.org/10.1016/j.cell.2022.12.018 (2023).
BRASIL. Ministério da Saúde. Gabinete do Ministro. Nota Técnica nº 1217, de 17 de setembro de 2021. https://www.gov.br/saude/pt-br/coronavirus/notas-tecnicas/2021/sei_ms-0023072995-nota-tecnica_plano-de-expansao-da-testagem.pdf/view
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34(18), 3094–3100. https://doi.org/10.1093/bioinformatics/bty191 (2018).
Aksamentov, I., Roemer, C., Hodcroft, E. B. & Neher, R. A. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 6(67), 3773. https://doi.org/10.21105/joss.03773 (2021).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12), 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 (2012).
Singh, P., Sharma, K., Shaw, D., Bhargava, A. & Negi, S. S. Mosaic recombination inflicted various SARS-CoV-2 lineages to emerge into novel virus variants: A review update. Ind J Clin Biochem 38(4), 1–8. https://doi.org/10.1007/s12291-022-01109-w (2022).
Moreira, E. D. Jr. et al. Safety and efficacy of a third dose of BNT162b2 covid-19 vaccine. N. Engl. J. Med. 386(20), 1910–1921. https://doi.org/10.1056/NEJMoa2200674 (2022).
Lustig, Y. et al. Superior immunogenicity and effectiveness of the third compared to the second BNT162b2 vaccine dose. Nat. Immunol. 23(6), 940–946. https://doi.org/10.1038/s41590-022-01212-3 (2022).
Collie, S. et al. Effectiveness and durability of the BNT162b2 vaccine against Omicron Sublineages in South Africa. N. Engl. J. Med. 387(14), 1332–1333. https://doi.org/10.1056/NEJMc2210093 (2022).
Arunachalam, P. S. et al. Durability of immune responses to mRNA booster vaccination against COVID-19. J. Clin. Invest. 133(10), e167955. https://doi.org/10.1172/JCI167955 (2023).
Tay, M. Z. et al. Heterologous mRNA vaccine boosters induce a stronger and longer-lasting antibody response against Omicron XBB variant. Lancet Reg. Health West Pac. 33, 100732. https://doi.org/10.1016/j.lanwpc.2023.100732 (2023).
Vicentini, M. et al. Risk of SARS-CoV-2 reinfection by vaccination status, predominant variant and time from prior infection: A cohort study, Reggio Emilia province, Italy, February 2020 to February 2022. Euro Surveill. 28(13), 2200494. https://doi.org/10.2807/1560-7917.ES.2023.28.13.2200494 (2023).
Cele, S. et al. SARS-CoV-2 prolonged infection during advanced HIV disease evolves extensive immune escape. Cell Host. Microbe. 30(2), 154-162.e5. https://doi.org/10.1016/j.chom.2022.01.005 (2022).
Dan, J. M. et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 371(6529), eabf4063. https://doi.org/10.1126/science.abf4063 (2002).
Turner, J. S. et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 596(7870), 109–113. https://doi.org/10.1038/s41586-021-03738-2 (2021).
Dawood, R. M. et al. Bioinformatics prediction of B and T cell epitopes within the spike and nucleocapsid proteins of SARS-CoV2. J. Infect. Public Health 14(2), 169–178. https://doi.org/10.1016/j.jiph.2020.12.006 (2021).
Qu, P. et al. Enhanced neutralization resistance of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host. Microbe. 31(1), 9–17. https://doi.org/10.1016/j.chom.2022.11.012 (2023).
Ridgway, H. et al. Molecular epidemiology of SARS-CoV-2: The dominant role of arginine in mutations and infectivity. Viruses 15(2), 309. https://doi.org/10.3390/v15020309 (2023).
Alcantara, M. C., Higuchi, Y., Kirita, Y., Matoba, S. & Hoshino, A. Deep mutational scanning to predict escape from bebtelovimab in SARS-CoV-2 omicron subvariants. Vaccines (Basel) 11(3), 711. https://doi.org/10.3390/vaccines11030711 (2023).
Qu, P. et al. Enhanced evasion of neutralizing antibody response by Omicron XBB15, CH11, and CA31 variants. Cell Rep. 42(5), 112443. https://doi.org/10.1016/j.celrep.2023.112443 (2023).
Ao, D., He, X., Hong, W. & Wei, X. The rapid rise of SARS-CoV-2 Omicron subvariants with immune evasion properties: XBB.1.5 and BQ.1.1 subvariants. MedComm (2020) 4(2), e239. https://doi.org/10.1002/mco2.239 (2023).
Guo, H. et al. Increased resistance of SARS-CoV-2 Lambda variant to antibody neutralization. J. Clin. Virol. 150–151, 105162. https://doi.org/10.1016/j.jcv.2022.105162 (2022).
Wang, M. et al. Reduced sensitivity of the SARS-CoV-2 Lambda variant to monoclonal antibodies and neutralizing antibodies induced by infection and vaccination. Emerg. Microbes. Infect. 11(1), 18–29. https://doi.org/10.1080/22221751.2021.2008775 (2022).
Wang, X. et al. SARS-CoV-2 ORF8 protein induces endoplasmic reticulum stress-like responses and facilitates virus replication by triggering calnexin: An unbiased study. J. Virol. 97(3), e0001123. https://doi.org/10.1128/jvi.00011-23 (2023).
Lin, X. et al. Unconventional secretion of unglycosylated ORF8 is critical for the cytokine storm during SARS-CoV-2 infection. PLoS Pathog. 19(1), e1011128. https://doi.org/10.1371/journal.ppat.1011128 (2023).
Rogozin, I. B., Saura, A., Bykova, A., Brover, V. & Yurchenko, V. Deletions across the SARS-CoV-2 genome: Molecular mechanisms and putative functional consequences of deletions in accessory genes. Microorganisms 11(1), 229. https://doi.org/10.3390/microorganisms11010229 (2023).
Hassan, S. S. et al. A unique view of SARS-CoV-2 through the lens of ORF8 protein. Comput. Biol. Med. 133, 104380. https://doi.org/10.1016/j.compbiomed.2021.104380 (2021).
Ortiz-Pineda, P. A. & Sierra-Torres, C. H. Evolutionary traits and genomic surveillance of SARS-CoV-2 in South America. Glob. Health Epidemiol. Genom. 2022, 8551576. https://doi.org/10.1155/2022/8551576 (2022).
Menezes, D., Fonseca, P. L. C., de Araújo, J. L. F. & Souza, R. P. SARS-CoV-2 genomic surveillance in Brazil: A systematic review with scientometric analysis. Viruses 14(12), 2715. https://doi.org/10.3390/v14122715 (2022).
Acknowledgements
The authors would like to thank all the participants in this study, the Universidade Federal de Santa Maria, the Secretaria de Município da Saúde de Santa Maria, and the financial support of the Brazilians' development agencies.
Funding
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES); Ministério da Ciência, Tecnologia e Inovações (MCTI); and Bill e Melinda Gates Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualization: B.C.P., L.F.T., and P.A.T.; Methodology: B.C.P., T.R.yC., L.F.T., B.C.C., A.A.V.; Formal analysis: B.C.P., L.F.T., A.A.V.; B.C.C.; Investigation: T.R.yC., B.C.C., V.T.S., A.P.S., and P.A.T.; Resources: A.V.S., and P.A.T.; Data curation: B.C.P., L.F.T., A.A.V.; Writing—original draft preparation: B.C.P., L.F.T., B.C.C., and T.R.yC.; Writing—review and editing: A.V.S. and P.A.T.; Visualization: B.C.P., and L.F.T.; Supervision: P.A.T.; Project administration: A.V.S. and P.A.T.; Funding acquisition: A.V.S. and P.A.T. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Piccoli, B.C., y Castro, T.R., Tessele, L.F. et al. Genomic surveillance and vaccine response to the dominant SARS-CoV-2 XBB lineage in Rio Grande do Sul. Sci Rep 14, 16831 (2024). https://doi.org/10.1038/s41598-024-67828-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-67828-7
