
Abstract
Coral microbiomes play crucial roles in holobiont homeostasis and adaptation. The host’s ability to populate broad ecological niches and to cope with environmental changes seems to be related to the flexibility of the coral microbiome. By means of high-throughput DNA sequencing we characterized simultaneously both bacterial (16S rRNA) and Symbiodiniaceae (ITS2) communities of four reef-building coral species (Mussismilia braziliensis, Mussismilia harttii, Montastraea cavernosa, and Favia gravida) that differ in geographic distribution and niche specificity. Samples were collected in a marginal reef system (Abrolhos, Brazil) in four sites of contrasting irradiance and turbidity. Biological filters governed by the host are important in shaping corals’ microbiome structure. More structured associated microbial communities by reef site tend to occur in coral species with broader geographic and depth ranges, especially for Symbiodiniaceae, whereas the endemic and habitat-specialist host, M. braziliensis, has relatively more homogenous bacterial communities with more exclusive members. Our findings lend credence to the hypothesis that higher microbiome flexibility renders corals more adaptable to diverse environments, a trend that should be investigated in more hosts and reef areas.
Similar content being viewed by others


Introduction
Corals are holobionts, a complex consortium composed of the cnidarian host and its associated microbiome1. The corals’ association with dinoflagellates of the family Symbiodiniaceae provides energetic support to the cnidarians2. Physiological characteristics of specific Symbiodiniaceae lineages3 influence holobiont’s performance, stability, and resilience4,5. Bacteria are concentrated in coral holobionts compared to the surrounding seawater, benefiting their host through mechanisms such as photosynthesis, protection against infection, nutrient cycling and provision. For example, some taxa are recognised as potential metabolizers of organic sulfur compounds, such as Alteromonas and Pseudomonas6, and as nitrogen fixers, such as Cyanobacteria7. Conversely, opportunistic bacteria may surpass their community neighbors and cause dysbiosis or disease when corals are stressed1,6. Despite the vast overall diversity, three subcommunities have been characterized in corals, one responsive to abiotic and biotic drivers across spatial and temporal scales, regional core phylotypes filling functional niches, and a host-selected, persistent core microbiome8.
Symbiodiniaceae and bacteria community structure strongly depend on the coral species4,9,10,11,12,13, which range from specialist to generalist regarding associations with their microbiome10,14. Host microbial flexibility—the ability to harbor different microbiomes—in generalist taxa may be influenced by many drivers. The environmental factors (e.g., temperature, nutrients, latitude) can favor some taxa as thermotolerant photosymbionts15 or copiotrophic bacteria16. The acquisition mode of Symbiodiniaceae cells14,17 demonstrated that the horizontal transmitter provides numerous Symbiodiniaceae taxa involved in community composition, which could influence the flexibility of the community14,18. Additionally, genetic divergence within the host population19,20 positively correlated with the structuring of symbiotic communities. Stochastic events that disturb local host-microbiome associations e.g., heat waves and storms, might lead to higher variability in communities21. Besides the listed factors, biotic interactions can also affect diversity and influence adaptation to the environment22. Microorganisms in complex communities, such as holobionts, engage in numerous ecological interaction webs that shape the structure and function of these communities. These interactions might have positive (e.g., metabolic cooperation), negative (e.g., predation) or no impact on the species involved, including the host23.
Shifts in microbiome structure have been argued to be an efficient adaptation mechanism in sessile corals1,24. A comprehensive discussion about the mechanisms and tradeoffs of microbiome structures (specific vs. flexible) has been conducted by Voolstra and Ziegler25. They propose that while higher microbiome flexibility should promote fast responses to environmental changes, it makes more unstable communities prone to opportunistic infections. Conversely, lower flexibility should support stable and efficient associations but reduces adaptation capacity. While this model is primarily based on temporal changes over the same organism, it has already been hypothesized that the same rationale could be applied to the heterogeneity of ecosystems14,18,26. For instance, coral species with wider depth ranges, so-called “depth generalists”, tend to exhibit higher symbiont zonation—i.e. specific symbiont taxa occurring at specific depth intervals—than those with narrower depth ranges, or “depth-specialists”18. The number of distinct Symbiodiniaceae lineages observed in association with the corals was positively correlated with coral species’ geographic distribution14. Additionally, corals that obtain Symbiodiniaceae from the environment had more levels of symbiont community zonation18. The variability of the bacterial community also appears to follow these trends as depth generalist corals had more variable bacterial communities characteristic of each depth range26. They propose that this association of corals with different symbiont taxa throughout depth contributes to the host’s wider distribution18,26.
The variability of coral microbiomes as a response to environmental conditions has been addressed for both Symbiodiniaceae and bacteria. These components of the microbiome show specific responses to environmental and host differences13,15,27,28,29,30. The bacterial community is likely to have more complex dynamics than Symbiodiniaceae as the former is much more diverse and inhabit various hosts compartments such as mucus, tissue and the endolytic space1 whereas the latter is restricted to the gastrodermis2. However, comparative studies describing both bacteria and Symbiodiniaceae diversity in more than one host species are scarce. Claar et al.29 suggests higher variability for both communities in reefs highly impacted by human disturbances. Furthermore, disturbance was consistently associated with higher bacterial diversity and richness whereas no such association was observed in Symbiodiniaceae. Another study11 reports the host species as one of the underlying factors influencing the structure and composition of Symbiodiniaceae communities as well as a strong driver of coral-associated bacterial community composition.
We hereby assessed bacterial and Symbiodiniaceae communities structure in four coral host species of contrasting biological traits on a regional scale in the largest (46.000 km2) and more diverse reef system of the Southwestern Atlantic Ocean, the Abrolhos Bank, off the Brazilian coast. Corals were sampled in sites of contrasting environmental conditions. We address the hypothesis that widely distributed and depth-generalist hosts have more variable microbiomes influenced by environmental conditions than endemic and depth-specialist hosts. For that we explored associations among the Symbiodiniaceae and bacterial components by analyzing them simultaneously in a set of coral specimens. This provided a concise and robust framework for investigating bacteria-Symbiodinaceae community structure and their responses to biotic and abiotic conditions. Our results comprise the first broad characterization of coral-associated Symbiodiniaceae and bacterial community cooccurrence in turbid-zone reefs.
Materials and methods
Organisms and sampling
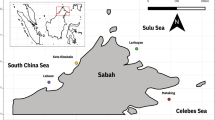
Four coral species of different depth and geographical ranges, Montastraea cavernosa (Linnaeus, 1766), Mussismilia braziliensis (Verrill, 1868), Mussismilia harttii (Verrill, 1902) and Favia gravida (Verrill, 1868), were sampled in the Abrolhos Bank, Brazil, in the tropical South Atlantic Ocean (SAO) (Fig. 1, Supplementary Information S1). Montastraea cavernosa is a broadcast spawner with asymbiotic larvae and occurs in the Caribbean and the tropical and subtropical SAO (Brazilian coast). This species occurs from near surface to a depth of 113 m, at higher abundance and having larger size in low-light environments31,32. Mussismilia braziliensis and M. harttii are also broadcast spawners with asymbiotic larvae but are endemic to the western SAO. The former occurs only from ca. 10° S to ca. 20° S, mostly in shallow (0–25 m), well-lit habitats33,34,35, whereas M. harttii occurs from ca. 5° to 20° S, between 0 and 50 m depths36,37. Favia gravida is a brooder with internal fecundation that releases planula containing Symbiodiniaceae cells. It is also broadly distributed on both sides of the SAO (West Africa and Brazil) and oceanic islands32,38, ranging from ca. 19° N to 20° S. It thrives in well-lit and shallow (0–30 m) habitats and intertidal pools. Additionally, Mussismilia braziliensis and Montastraea cavernosa are dominant species in the reef tops and walls, respectively. Together, they represent most of Abrolhos’ coral cover33. Favia gravida, M. harttii and M. braziliensis only occur in tops, while M. cavernosa occurs at both reef tops and walls33.

Top: Geographic distribution of the investigated coral species according to IUCN (www.iucnredlist.org) and occurrences registered in Brazil20. Colors correspond to species in the photographic inserts (bottom right panel). The black square indicates the Abrolhos Bank, off Bahia (BA) and Espírito Santo (ES) states, Brazil. Bottom left: Location of the four sampling sites (TIM Timbebas, SG Sebastião Gomes, PAB Parcel dos Abrolhos, ESQ Esquecidos). Colors represent the coral species sampled at each site. Red, dashed polygons indicate Abrolhos National Marine Park, a no-take Protected Area. Gray lines and numbers are the depth contours. Bottom right insert: Coral species in this study; colors match the distributions in the upper panel. Photographs: Livia Villela and Felipe Ribeiro.
Samples and environmental data were collected at four sites (50 to 170 km apart) with contrasting characteristics: TIM: Timbebas, SG: Sebastião Gomes, PAB: Parcel dos Abrolhos, and ESQ: Esquecidos (Fig. 1, Supplementary Information S1). The coastal reef (Timbebas, TIM, pinnacles tops at 5–8 m deep) lies within a poorly enforced portion of the Abrolhos National Marine Park (ANMP). It shows slightly lower turbidity than the other coastal reefs located southward. The coastal reef Sebastião Gomes (SG, pinnacles tops at 1–6 m deep) is closer to the coast and slightly shallower than TIM, subjected to higher turbidity and sedimentation from a dredging disposal area. The most offshore reef (Parcel dos Abrolhos, PAB, pinnacles tops at 7–12 m deep) is located near the Abrolhos Archipelago and within the well-enforced portion of the ANMP. The southernmost sampling site (Esquecidos, ESQ, pinnacles tops at 15–20 m depth) is also the deepest and closest to the Doce River mouth, being more influenced by its plume. Five specimens of each coral species were collected by SCUBA diving at sites PAB, SG, TIM and ESQ. At site ESQ, only M. cavernosa and M. harttii were collected (M. braziliensis was absent in the region, and F. gravida was not found in the sampled pinnacle, despite being present on other pinnacles in the region). Samples were collected by detaching a whole coral colony or breaking off small pieces of large colonies with chisel and hammer. At each site, samples were collected at similar depths, with colonies being of similar size (ca. 5–10 cm in diameter), polyps (2 to 4) in the case of M. harttii, or a fragment of large colonies as M. cavernosa. Colonies were photographed in situ using a color reference palette and scale before sampling. Once onboard, the samples were kept in seawater inside individual plastic bags for 1–2 h and tissue was extracted with a pressurized air blow gun and homogenized with 15 mL of 0.22 µm filtered seawater; the final volume was registered and preserved in 2 mL aliquots in liquid nitrogen.
DNA extraction, amplification and sequencing
DNA was extracted from tissue homogenates of each coral specimen according to Fukami et al.39. Briefly, the coral tissue was lysed in a chaotropic saline buffer for 1 week, and the DNA was extracted with phenol–chloroform-isopropanol followed by 75% ethanol rinses. The DNA concentration was adjusted to 15 ng/μL with Milli-Q water and purified with a ProNex® size-selective purification system following the manufacturer’s instructions.
Symbiodiniaceae communities were studied by sequencing the internal transcribed spacer II region (ITS2). Amplification was performed following an improved primer set (Sym_VAR_5.8SF/Sym_VAR_REV) with Illumina adapter tails tested by Hume et al.40 and using Qiagen hot start High-fidelity Taq polymerase. The PCR program consisted of an initial denaturation at 95 °C (5 min), followed by 35 cycles of denaturation at 95 °C (30 s), annealing at 56 °C (30 s) and extension at 72 °C (40 s), and a final extension of 72 °C (5 min). PCR products were checked for size using gel electrophoresis and quantified in a NanoDrop Lite spectrophotometer. Sequencing was performed on an Illumina MiSeq platform with an average of 50,000 paired-end 250 bp-long reads per sample.
Bacterial communities were investigated by sequencing 16S rRNA gene amplicons. The primers 341F and 785R were used to amplify the V3–V4 regions of the 16S rRNA gene41. The reaction mixture (50 μL total volume per sample) consisted of Econotaq® PLUS GREEN 2X Master Mix (Lucigen) (25 μL), Ambion® nuclease-free water (17 μL), the primer pair 341F and 785R (1.5 μL of each; 10 μM) and DNA template (5 μL). The PCR program consisted of an initial denaturation at 94 °C (2 min), followed by 35 cycles of denaturation at 94 °C (30 s), annealing at 55 °C (30 s) and extension at 72 °C (40 s), and a final extension of 72 °C (7 min). PCR products were quantified using gel electrophoresis. 16S rRNA gene amplicon sequencing was performed on an Illumina MiSeq platform with an average of 100,000 paired-end 300 bp-long reads per sample.
Sequence processing and bioinformatic analysis
The bioinformatic processing of ITS2 sequences was performed using the analytical framework implemented by Symportal42. Sequences with 100% identity received the known name (e.g., A4) given in the Symportal database, while unknown sequences received the clade/genus code (A to I) followed by 4 random digits (e.g. C1434).
The transcribed internal spacers of the rRNA gene are the most commonly used region to infer Symbiodiniaceae diversity, allowing intragenus resolution (a.k.a. ITS2 types), but also present multiple copies with intragenomic variants denominated as IGVs43. One widely accepted premise is that the most frequent copy of the genome characterizes the sequence of ITS2 types43. Our approach consisted of capturing the most prevalent ITS2 sequences to minimize the probability of contaminating the final ITS2 type dataset with IGVs. We used each genus’s 20% most frequent ITS2 sequences to produce two pairwise correlation matrices (Pearson), one with 58 Cladocopium sequences and 32 Symbiodinium sequences. Correlations among ITS2 sequences were calculated with the package ggcorrplot in R, with sequences clustered by absolute hierarchical clustering. Only the most abundant sequence in each cluster (positive gamma correlation) was considered a valid ITS2 type. This conservative approach was a tradeoff between missing some low-frequency ITS2 types and increasing the chance of unveiling new abundant ones while reducing the probability of assigning intragenomic copies to the ITS2 type status. Statistical analyses (nMDS and PERMANOVA, see Community analysis) were performed on both datasets (ITS2 total sequences and types) to check for the effect of complexity reduction on community structure. The relative abundance of Symbiodiniaceae ITS2 types was estimated for each of the 64 coral specimens separately, based on a dataset normalized by the one with the lowest number of reads (see Rarefaction curves). Haplotype networks were made separately for each Symbiodiniaceae genus using the pegas R package to reconstruct genetic relations among ITS2 types. Networks were edited to include information on indels or SNPs—single nucleotide polymorphisms.
Sequence data of the 16S rRNA gene were initially quality‐filtered and trimmed using Trimmomatic version 0.3644; truncating reads if the quality dropped below 20 in a sliding window of 4 bp. USEARCH version 11.0.66745 was used for further processing. Sequences were denoised and clustered into unique sequences (zero-radius operational taxonomic units; zOTUs) using the UNOISE algorithm implemented in USEARCH. Chimeric sequences were removed with UCHIME de novo during zOTU clustering and subsequently with a reference-based comparison against the genome taxonomy database (GTDB) (https://gtdb.ecogenomic.org/). zOTUs were taxonomically classified using a Bayesian Last Common Ancestor algorithm (BLCA) against the GTDB. Nonbacterial OTUs were removed along with singleton OTUs. Finally, processed sequences were mapped on zOTU sequences to calculate the count distribution and counts of each zOTU in every sample. Only zOTUs occurring in more than two samples were considered for further statistical analysis. The classification of the core microbiome (refer to next section) zOTUs shared by all hosts (n = 10) was further investigated using SINA aligner (v1.2.12) and the taxonomies hosted by the SILVA database. A maximum-likelihood tree was built including the ten closest neighbors of each zOTU using FastTree46.
Biodiversity estimation
The number of sequences was standardized across samples to account for different sequencing depths by randomly subsampling each sample to the lowest number of sequences (i.e. 19,407 and 9856 counts for bacterial and Symbiodiniaceae community data, respectively). Rarefaction curves based on rarefied data and Good’s coverage were calculated in R 3.5.347 with the vegan package48. Richness and Shannon’s index to ITS2 total sequences, ITS2 types and zOTU were calculated. The Kruskal–Wallis rank sum test, followed by Wilcoxon’s pairwise comparisons, was used to evaluate differences in richness and alpha diversity among different groups. A p-value < 0.05 was considered significant after corrections for multiple comparisons49. The statistical comparisons of alpha-diversity and data dispersion by reef were restricted to reefs with all species sampled (PAB, SG and TIM) and to all reefs with only the two coral species sampled at all (M. harttii and M. cavernosa). The comparisons by species were made without ESQ site, to avoid misinterpretation of the data due to an uneven number of samples. Even so, the graphics demonstrated all the data together. The number of zOTUs across at least one sample from each group (host taxon or reef site) and the shared associations in all categories were detected and plotted as Venn diagrams. To infer the “core microbiome”, we used a presence in at least 80% of samples for each category.
Community analysis
Dissimilarities in the bacterial (zOTU) and Symbiodiniaceae communities (ITS2 types and ITS2 total sequences) were calculated with the Bray–Curtis coefficient using square-root transformed abundance and presence-absence datasets. Dispersion of datasets was calculated with PERMDISP using the vegan package, comparisons between average distances from centroid were performed with Tukey honestly significant difference (HDS) and Principal Coordinate Analyses (PCoA) were performed. The effects of the factors “host species taxa”, “reef site”, “inshore/offshore reef”, “coral genus” and “host reproductive strategy” on community structure were tested separately and combined using permutational analysis of variance (PERMANOVA) with the distance matrices of abundance data. The same matrices were plotted in nonmetric multidimensional scaling (nMDS) optimized by the function sammon with the mass R package50. Additionally, significant factors were pairwise compared with 9999 random permutations using the pairwiseAdonis package. Environmental variables (depth, turbidity, temperature, optical depth) were contrasted against the Bray–Curtis distance matrices, and significant ones were added to the nMDS plot. Distance-based redundancy analysis was used to model the relationship between community structure and environmental predictors. A Bray–Curtis squared rooted matrix and the environmental variables (in situ temperature, turbidity, irradiance, depth and optical depth) were used in these analyses.
Indicator taxa of Symbiodiniaceae and bacteria were inferred using the function IndVal.g in the R package indicspecies51 using ITS2 type and zOTU data, respectively. The indicator species analysis determines which taxa are related to specific groups. This index consists of the probability to find the taxa in the sampling that belongs to a group, given that the taxa is found there (specificity or positive predictive value); and the probability of finding the species in sampling belonging to the evaluated group (fidelity or sensitivity). The final index IndVal ranges from 0 to 1. Taxa with higher specificity and fidelity were close to 1, and significance was tested by bootstraps. For each dataset, we searched independently for indicator taxa by coral species. Comparisons of ITS2 type and zOTU occurrence were made within each group (specimens of a given species or site) and between specimens and the remaining dataset outside the group.
Network analysis of cooccurrences and mutual exclusions of bacteria and Symbiodiniaceae was performed to investigate microbial interactions that may be relevant in structuring the communities. Datasets were pre-processed by retaining only bacterial zOTUs and Symbiodiniaceae ITS2 types with > 100 counts and present in > 1 sample, resulting in a final dataset with 62 samples, 1976 bacterial zOTUs and all 12 Symbiodiniaceae ITS2 types. The networks were built using normalized counts from the sequencing data of zOTUs and Symbiodiniaceae ITS2 types, with the latest version of SparCC52,53 implemented in FastSpar, with unbiased p-value estimator54. Analysis was made with 1000 iterations/1000 bootstraps. Associations with correlation scores >|0.5| and p values < 0.01 were considered for interpretation. Networks were analyzed with Cytoscape version 3.8.255. General properties were determined with Network Analyzer56, and clustering was calculated with ClusterMaker2 (www.rbvi.ucsf.edu/cytoscape/ClusterMaker2) using the Markov Clustering Algorithm (MCL) with the inflation value set to 2.0.
Ethics approval and consent to participate
Sampling permits: NGI Abrolhos/ICMBio 65055-8.
Results
Symbiodiniaceae and bacterial community data were obtained from 64 and 67 coral specimens, respectively, from 70 samples (Supplementary Information S2). A total of 537 distinct Symbiodiniaceae ITS2 sequences were identified after rarefaction, resulting in 12 unique ITS2 types. The rarefied bacterial 16S rRNA gene dataset generated 5,996,131 sequences, yielding 7114 unique zOTUs. Good’s 99.84% and 100% coverage revealed that most bacterial communities and ITS2 sequences were recovered, respectively (see rarefaction curves in Supplementary Information S4).
Symbiodiniaceae community structure
The most diverse and abundant ITS2 types were assigned to the genus Cladocopium (N = 7), followed by Symbiodinium (N = 3), with lower abundance and fewer occurrences of Breviolum and Gerakladium. Five undescribed ITS2 types (A1392, C1372, C1396, C1434, C1506) were retrieved (Supplementary Information S3). ITS2 type C3 was present in all coral specimens and was mostly dominant, except in M. harttii, where C3b and C1434 dominated. Other Cladocopium ITS2 types were detected in lower proportions (Fig. 2a). Symbiodinium was less diverse, with A4 being the most frequent. The unclassified ITS2 type A1392 was observed in three of the four coral hosts, and type A3 appeared in low proportions, albeit consistently, in M. cavernosa from reef site PAB. Richness was significantly lower in F. gravida (3.9, sd:1.1) than in other hosts (5.1–5.6) (Wilcoxon’s pairwise comparisons), especially in sites TIM (3.4, sd:0.5) and SG (3.6, sd:0.5) (p < 0.05) (Fig. 2b, Supplementary Information S4). Shannon’s diversity values were similar across sites and hosts.

(a) Composition of Symbiodiniaceae communities within coral microbiomes in the Abrolhos Bank: the host species are discriminated at the top of the plot, and the four reef sites are indicated at the bottom. (b) Richness and Shannon index; letters and symbols (*) indicate significant Wilcoxon’s pairwise comparisons test (p < 0.05 after BH correction). Tests and boxplot size were calculated without ESQ samples however their values were indicated in plots by dots. (c) Nonmetric multidimensional scaling (nMDS) plots calculated according to Bray–Curtis distance on the square root of the relative ITS2 type abundances. The stress of each nMDS is indicated in the figures. Colors correspond to host species taxa (FAGR Favia gravida, MOCA Montastraea cavernosa, MUBR Mussismilia braziliensis, MUHA Mussismilia harttii) and symbols to reef sites (SG Sebastião Gomes, PAB Parcel dos Abrolhos, TIM Timbebas, ESQ Esquecidos Reef).
Variability of Symbiodiniaceae beta-diversity in the relative abundance dataset was significantly lower for F. gravida (0.07) than for other hosts (0.28–0.33, p < 0.05—Tukey HSD). In contrast, in the occurrence dataset, it was higher in M. cavernosa (0.28) than in other coral species (0.13–0.18, p < 0.05—Tukey HSD) (Fig. 2c; Supplementary Information S5b,c). Community structure was related to coral host species (PERMANOVA; R2: 0.33; F.model: 9.83; p < 0.001). Reef sites (R2: 0.08; F.model: 4.2; p < 0.001) and the interaction between the host and each reef site (R2: 0.29; F.model: 6.81; p < 0.001) were also significant in explaining additional variance. “host reproductive strategy” (R2: 0.1; F.model: 7.1; p < 0.001) and “coral genus” (R2: 0.19; F.model: 7.3; p < 0.001) had significant influence on symbiont community structure when tested alone. However, they were not significant when analyzed together with host species. Host species, reef site and their interaction accounted for 70% of the variance in the data. The most different communities were those of F. gravida and Mussismilia species (PERMANOVA; R2: 0.47–0.32, p < 0.05) (Supplementary Information S5a). Significant differences were detected between M. braziliensis and M. harttii (R2: 0.24). Reef site-level differences were detected (p < 0.05) in the pairwise comparisons with compositional data between ESQ vs TIM (R2: 0.24), for presence/absence data to ESQ vs PAB (R2: 0.23), and for each host species separately (Fig. 3, Supplementary Information S6). Communities from M. cavernosa differed among sites (global: R2: 0.82; F.model 23.9; p < 0.001). The global PERMANOVA for F. gravida showed significant spatial variation (R2: 0.78; F.model 15.9; p < 0.001), but pairwise comparison did not show differences between the two coastal reefs (TIM vs SG), and comparisons with PAB were not possible due to the low number of samples from this site. M. harttii also had significant between-site differences (R2: 0.56; F.model 5.6; p < 0.001) but with lower structure than M. cavernosa and F. gravida. M. braziliensis had no detectable divergences between sites (R2: 0.13; F.model 0.8; p > 0.05) (Fig. 3, Supplementary Information S6). The nMDS and PERMANOVA with the complete ITS2 sequence dataset showed R2 and p-value similar to those obtained with the ITS2 type data (Supplementary Information S7).

Nonmetric multidimensional scaling (nMDS) discriminated by coral host species taxa showing dissimilarities within Symbiodiniaceae (left column) and Bacteria (right column) (Bray‒Curtis distance, square root transformed relative zOTU and ITS2 type abundances). R2, F.model values, and p significance (**p < 0.001) of the global PERMANOVAs are shown in each plot.
The analysis indicator taxa showed that Symbiodinium A4 was associated with M. braziliensis (IndVal index: 0.79), Cladocopium C1396 was associated with M. cavernosa (IndVal index: 0.73), and Cladocopium types C1434 (IndVal index: 0.84) and C3b (IndVal index: 0.71) were associated with M. harttii. No indicator taxa were detected in spatial patterns.
Bacterial community structure
Proteobacteria accounted for 57% of the total reads, with Vibrio, Sphingomonas, Photobacterium, and Ruegeria being the most dominant genera (Fig. 4a). A strain related to Sphingomonas sp. 000797515 (44% BCLA support) was present in all 64 samples and represented up to 11 and 3.2% of the relative and overall read abundance in single samples, respectively. Genera Sphingomonas and Ruegeria (phylum Proteobacteria), Rubritalea (Verrucomicribiota), and placeholder taxon UBA3125 (Actinobacteriota) were common to all samples.

(a) Composition of bacterial communities within coral microbiomes in Abrolhos Bank: host coral taxa and sampling sites are indicated. Bars represent the community composition of each coral specimen; color bars indicate the most abundant bacterial genera and grayscale bars represent zOTUs grouped by phylum. (b) Boxplots of richness (top) and Shannon diversity index (bottom) of the bacterial community discriminated by host species taxon (left) and reef site (right). Letters indicate significant differences (Wilcoxon’s pairwise comparisons test, p < 0.05 after BH correction). Tests and boxplot size were calculated without ESQ samples to Species measures and only by MOCA and MUHA to site measures, however, their values were indicated in plots by dots. (c) Nonmetric multidimensional scaling (nMDS) diagram of dissimilarities within communities (Bray–Curtis distance, square root transformed relative zOTU abundances). Colors correspond to host species (FAGR Favia gravida, MOCA Montastraea cavernosa, MUBR Mussismilia braziliensis, MUHA Mussismilia harttii) and symbols to reef sites (SG Sebastião Gomes, PAB Parcel dos Abrolhos, TIM Timbebas, ESQ Esquecidos Reef).
Average richness was significantly lower (Wilcoxon’s pairwise comparisons) in ESQ (240.0, sd: 126.3) than in SG and TIM (702.0, sd: 336.1 and 477.5, sd: 244.3, respectively) (Fig. 4b, Supplementary Information S4). Data dispersion was not significantly different between groups of species and reef sites (Supplementary Information S8b,c). The PERMANOVAs with abundances’ data demonstrated that reef sites (R2: 0.14; F.model: 3.52; p < 0.001) and host species (R2: 0.13; F.model 3.16; p < 0.001), independently, were significantly related with the bacterial community structure (Fig. 4c). The interaction between host species and reef sites was significant and explained an additional variance in the data (R2: 0.21; F.model: 3.02; p < 0.05). Host species, reef site, and their interaction explained 47% of the variability. The other factors, “inshore/offshore reef”, “coral genus” and “host reproductive strategy”, were not significant in the model with host species and reef site. Low but significant (p < 0.05) R2 values were observed in all most pairwise comparisons among sites or host species, from 0.07 to 0.19 (Supplementary Information S8a). The divergence among reef sites for each host species (Fig. 3, Supplementary Information S6) was significant for all species and with a high to low effect from F. gravida (global R2: 0.52; F.model: 6.44; p < 0.001), M. cavernosa (global: R2:0.43; F.model: 3.52; p < 0.001), M. harttii (global R2: 0.35; F.model: 2.74; p < 0.001) and M. braziliensis (global R2: 0.25; F.model: 2.0; p < 0.001). Turbidity, depth, optical depth (Kd490 * depth), and in situ temperature were significantly correlated with bacterial community structure (R2; temperature: 0.21, turbidity: 0.14, optical depth: 0.16, depth: 0.31) (Fig. 3c) and explained together 17.9% of the differences (dbRDA; p < 0.001) (Supplementary Information S9).
The indicator species among the 10% most abundant strains include the zOTU394 Photobacterium aphoticum (BCLA: 29.45) in F. gravida (IndVal score 0.75), and the zOTU1525 OLB17 sp003223025 (Pyrinomonadaceae) in M. braziliensis (IndVal score 0.72). On a reef site basis, the ESQ reef’s main indicators were zOTU6 (BK-30 sp002837145–BCLA: 38.51) (IndVal score 0.74). Most indicator taxa of the SG reef belonged to the Vibrionaceae family: zOTU31 P. aquae (BCLA: 66.42), zOTU54 Vibrio hyugaensis (BCLA: 28.67), and zOTU67 V. neptunius (BCLA: 54.87); and to the phylum Acidomicrobia (zOTU727 Bin76 sp002238785) with IndVal scores of 0.88, 0.79, 0.71, and 0.71, respectively (Supplementary Information S10).
Only 9.1% of the bacterial zOTUs were shared between the four sites. However, when excluding the less sampled site ESQ, shared zOTUs increased to 24.1% (Supplementary Information S11a). zOTUs common to all host species taxa represented 21.6% of all zOTUs, 30.8% were exclusive to single sites, and 26.5% were exclusive to a single host. The family Pirellulaceae was the most diverse taxon shared to all hosts, while Woeiseia oceani (Woeseiaceae) was the most frequent. The predominant zOTUs occurring in F. gravida were members of the family Vibrionaceae, while Flavobacteriaceae predominated in M. harttii, and Pirellulaceae predominated in M. cavernosa and M. braziliensis.
Using an 80% prevalence cutoff for core microbiome inference, the total number of zOTUs dropped from 7114 to only 105 when using host species taxa and to 90 when reef sites were used as a grouping condition (Fig. 5a,b). Core zOTUs shared by all hosts (10 zOTUs, Fig. 5a; Supplementary Information S11b,c) were mostly from phylum Proteobacteria, four belonging to the order Burkholderiales (Acidovorax_D, Achromobacter dolens, BK-30, and Ralstonia pickettii) and the remaining to genera Sphingomonas, Stenotrophomonas and Caulobacter. Other core zOTUs from all host taxa were classified as Synechococcus (Cyanobacteria) and Aeribacillus (Firmicutes). Six of ten zOTUs in the core microbiome shared by all hosts (Fig. 5a) were also present in all reef sites (Fig. 5b). Richness was much lower at site ESQ (1246 zOTUs—Fig. 5c). The host M. braziliensis and site TIM had the highest number of zOTUs classified as core (e.g. present in almost 80% of samples from this host/reef) (77 and 59, respectively) and exclusive core (50 zOTUs in M. braziliensis and 40 zOTUs in site TIM—e.g. present in almost 80% of samples from the host/reef, and not in the others host/reefs). Also, TIM had the higher proportion of zOTUs present in core (1.07%) (Fig. 5b,c).

Venn diagrams of the number and percentages of bacterial zOTUs among host species (a) and reef sites (b) using zOTUs present in at least 80% of the samples (n = 105 zOTUs considering host species as class, and n = 90 zOTUs considering reef site). (c) Total number of zOTUs, prevalent (> 80%) zOTUs per host species and reef site, percent of prevalent zOTUs in relation to the total number of zOTUs, and number of exclusive zOTUs per group. Sign w/o = without.
Microbial network
The network encompassed 1116 edges linking 228 nodes (5 Symbiodiniaceae ITS2 type and 223 bacterial zOTUs), with a clustering coefficient of 0.591, 3.4 average path length, 13.8 average number of neighbors, 0.098 density, and 0.525 modularity (Supplementary Information S11). The main module was composed of three interconnected clusters (Fig. 6). The larger cluster I (81 zOTUs) consisted predominantly of more connected Vibrionaceae (mostly Vibrio and Photobacterium). The top ten hubs (i.e. strongly connected nodes) of cluster I were classified as V. neptunius, V. tubiashii, V. ponticus, V. maritimus, V. sinaloensis, and P. aquae (Fig. 6). Cluster II (51 zOTUs) was dominated by Sphingomonas, Ralstonia, Firmicutes and Burkholderiales nodes. Negative correlations indicating mutual exclusions were observed among two nodes of Vibrionaceae cluster I (V. parahaemolyticus and V. tubiashii) and the Sphingomonas sp. 000797515 (BCLA 44%) node in cluster II, and also among V. parahaemolyticus and BK-30 (Burkholderiaceae) in cluster II. There was also a negative correlation between Deinococcus actinoscleris from cluster II and UBA5680, a node in cluster III.

Main microbial network module with three clusters (I, II, and III). Nodes are bacterial zOTUs (color-coded circles with area proportional to relative abundance) and one Symbiodiniaceae ITS2 type (octamer with proportional area). Lines linking the nodes (edges) represent positive (gray) or negative (red) correlations (FastSpar coefficient ≥ |0.5|, p-corrected value < 0.01). Edge thickness is proportional to the strength of the correlation between nodes, and thicker lines surrounding nodes indicate the network’s hubs. Bar plots represent the proportion of bacterial zOTUs within each cluster, grouped by host species (FAGR Favia gravida, MOCA Montastraea cavernosa, MUBR Mussismilia braziliensis, MUHA Mussismilia harttii) and reef site (SG Sebastião Gomes, PAB Parcel dos Abrolhos, TIM Timbebas, ESQ Esquecidos Reef). The entire network is presented in Supplementary Information S11.
Members of the Vibrionaceae cluster (I) were found in all sites and hosts, but predominantly at SG (69%) and in F. gravida (67%) (Fig. 6, cluster I, bar plot). Members of cluster II were more evenly distributed among sites, with M. harttii and M. cavernosa hosting the higher proportion of zOTUs and representing nearly 80% of the total (Fig. 6, cluster II). As expected, fewer connections involving Symbiodiniaceae ITS2 types were detected, given their lower number compared to the number of zOTUs. Two duplets connected Symbiodiniaceae nodes, ITS2 types A4 and C3 showed mutual exclusion and a cooccurrence linked A1392 with C8c (Supplementary Information S11). The only connection with bacterial zOTUs was positive and involved the novel ITS2 type C1434 and four bacterial zOTUs, the Sphingomonas sp. 000797515 (BCLA 44%) hub, and the Vulcanococcus, Bradyrhizobium, and Marinobacter nodes in cluster II. Another highly interconnected cluster (cluster IV, Supplementary Information S11), with zOTUs distributed among all coral species, was recorded in site TIM. Most of the cluster’s zOTUs (20) were from families Flavobacteriaceae (7) and Rhodobacteraceae (9). Otherwise, the network presented smaller (2 to 9 nodes) and peripheral clusters with 1 to 5 edges (Cluster III Fig. 6; complete network in Supplementary Information S11). Cluster III, with zOTUs interacting mainly in M. braziliensis, shared remarkably few zOTUs with clusters I and II. These sparse clusters also encompassed bacterial families that were less represented in the main clusters, such as Pirellulaceae, Rhodobacteraceae, Sedimentocolaceae and Verrucomicrobiaceae (Supplementary Information S11).
Discussion
Here, by assessing the bacterial and Symbiodinaceae microbiomes of corals in four contrasting reefs in the Abrolhos Bank, Brazil, we observed that corals with broader geographic distributions and depth ranges tend to harbor microbiome communities that exhibit higher dissimilarity between sites. This feature was somehow different between bacteria and Symbiodiniaceae, the latter displaying a stronger gradient in the effect of sites across hosts. In Symbiodiniaceae, the mode of acquisition of symbionts by the coral (broadcast vs. brooding) seems to influence symbiont community heterogeneity on a regional scale. Among all tested factors, the microbial community’s overall structure was better explained by host species for Symbiodiniaceae and by host and reef sites combined for bacteria. Both factors explained as much as 70% of the Symbiodiniaceae community structure. In contrast, more than half of the variance in the bacterial community dissimilarity (53%) was either stochastic or unexplained, which is not unexpected, since the coral holobiont encompasses a huge and diverse transient community compared to the more stable and selected core microbiome8, equivalent to 0.01% of the entire community in this study. On the other hand, biological interactions play significant roles in structuring microbial communities. Their characterization helps to decipher complex patterns in microbial profiles and detect the protagonist microbes of community engineering23. The microbial network revealed co-occurrences and mutual exclusions unevenly distributed in hosts and reefs, highlighting potentially protagonist microbes structuring the coral’s assemblages (3% of the overall bacteria and 42% of the photosymbionts). Furthermore, our results expanded the knowledge on coral symbiont diversity, as several undescribed taxa of both Symbiodiniaceae and Bacteria were detected. A putative relationship between host geographic distribution, host traits, and symbiotic microbiome characteristics is summarized in Fig. 7.

Summary of the main characteristics of the four coral species investigated in this study regarding their percentage of potentially pathogenic Vibrio taxa and microbial profiles. Bleaching susceptibility is based on Teixeira et al.77. Microbiome structure refers to the composition of the community, considering occurrence and relative abundance of taxa. Values in purple gradient refer to R2 values of global PERMANOVA analysis by host (Fig. 5) to Symbiodiniaceae and Bacteria, respectively.
Cladocopium (mainly C3) and Symbiodinium (mainly A4) occur at high frequencies in western SAO corals57,58,59, which is in line with their dominance in our samples. The occurrence and abundance of C3 (abundant in all hosts and sites) and A4 (undetectable in ESQ) and their pattern in the network suggests an antagonistic interaction between them. The undescribed ITS2 type C1434 was well distributed across hosts and particularly dominant in M. harttii at the ESQ site, which likely consists of a more suitable niche for this strain. Interestingly, this was the only ITS2 type positively correlated with bacteria in the network. The ITS type A3, associated with M. cavernosa, is a new occurrence for the region. The ITS2 type-based approach employed here agreed with results using the entire ITS2 sequence dataset. However, reducing the noise of intragenomic diversity by using the ITS2 type was critical to describe the most abundant taxonomic units, especially in a less-studied reef.
Host species significantly influenced the Symbiodiniaceae communities, and such associations seem to be driven by symbiont acquisition and the co-evolution between cnidarians and microalgae, along with the unique biochemical properties of cnidarian tissues60. Even so, the observed divergences in community composition were primarily due to the abundance of shared ITS2 types and not exclusive associations. While generalist symbionts may not render universal bleaching resistance to their hosts61, these redundant links could promote robustness62 by shifting through various potential symbionts to buffer oxidative stress during environmental disturbances63.
While pairwise comparisons among reef sites did not reveal significant differences, community variability (i.e. data dispersion) did differ. F. gravida, a brooder species that vertically transmits Symbiodiniaceae, displayed relatively low microbiome richness and beta diversity. A high community homogeneity is expected among spatially close coral colonies due to local recruitment17. It may be further amplified by well-adapted F. gravida Symbiodiniaceae strains established in parental colonies, transmitted during the planula phase. Despite that, this F. gravida is a generalist species with a broad amphi-Atlantic distribution59, and such traits appear to agree with the capacity to colonize contrasting environments such as coastal turbid reefs and oceanic islands.
The Brazilian endemic corals M. harttii and M. braziliensis presented lower and no significant dissimilarities of Symbiodiniaceae community among reef sites, respectively. The divergences in M. harttii are especially between the most distinct and distant site, ESQ, and the other reefs. Conversely to the other coral hosts studied herein, M. cavernosa exhibited the highest effect of reef site in variability of Symbiodiniaceae communities, which indicates more flexibility of the symbiotic community in response to environmental variation. This flexibility could be related to M. cavernosa wide geographical and bathymetric distribution along SAO and Caribbean environments31,62,64.
Proteobacteria (mostly Gammaproteobacteria and Alphaproteobacteria), Firmicutes, Actinobacteria and Bacteroidetes are usually associated with corals65. In the Abrolhos’ bacteriomes, Proteobacteria (currently Pseudomonadota) was the most abundant group, followed by Planctomycetota (mostly Pirellulaceae), Bacteroidota (includes former Bacteroidetes), Verrucomicrobiota and Actinobacteriota. Pirellulaceae and Woeseia, taxa that stood out in the coral’s common associations, have been reported in marine sediments66 and also associated with corals67. The high prevalence of sediment-characteristic taxa may stem from the high turbidity of the Abrolhos reefs68 (see Supplementary Information S1).
A proportionally small core microbiome agrees with the current understanding that coral’s harbor few conserved taxa8. Among bacteria present in all sites and hosts, we found taxa that are either common in coral microbiomes or less frequently reported but have the potential to structure these bacterial communities. For instance, Sphingomonas and Ralstonia occur intracellularly in Symbiodiniaceae69 and were the two most abundant and prevalent taxa in our coral microbiomes, closely relate to Sphingomonas sp. 000797515 and Ralstonia pickettii. Other taxa present in the core microbiome of all coral hosts included strains of Synechococcus, Burkholderiaceae, Acidovorax, Achromobacter, Stenotrophomonas and Aerobacillus. Ingestion of Synechococcus and other cyanobacteria cells by coral hosts contributes significantly to coral nutrition70. Synechococcus-like cyanobacteria have been extensively reported in association with coral tissues, where they play an essential role in nitrogen sourcing7. Aeribacillus pallidus is reported here for the first time in association with corals. This bacterium produces antimicrobial peptides71 and may play a role in the homeostasis of corals (e.g., by acting as a probiotic and shaping the host microbiome). It is worth to note that all core bacteria integrated the microbial network suggesting that these taxa indeed contribute to structure the communities of the hosts. Another noteworthy aspect was the scarcity of Endozoicomonas herein, which is in line with previous reports for Brazilian corals72,73,74,75. This contrasts with Endozoicomonas ubiquity in the Indo-Pacific and Caribbean, where it is an indicator of coral health65. It is possible that an alternative taxa—e.g. Sphingomonas for its abundance and ubiquity in the samples76—play analogous roles in the Abrolhos Bank.
The richness of the bacterial core and beta diversity patterns revealed contrasts between the specialist host M. braziliensis and the other coral species. M. braziliensis, with higher diversity and a higher percentage of core associations among all host species, is restricted to well-lit environments on pinnacles’ tops34 and might have developed a specialized microbiome. Specialist corals rely on stable microbial communities to cope with stressful environmental changes, avoiding reorganizing well-established symbioses12,25. Nevertheless, low flexibility does not necessarily reflect high vulnerability to environmental changes61. M. braziliensis showed a relatively low bleaching index (BI) during thermal anomalies in 201677 and 201978 although the much stronger event of 2019 was followed by higher mortality for this species than the 2016 one78,79.
The deeper ESQ site in the south, the closest to the Doce River mouth—and under the influence of its plume—showed the lowest richness than all the other sites (considering only M. harttii and M. cavernosa). The stressful conditions in this site could act as a selective pressure leading to the predominance of resistant and functionally important bacterial taxa while maintaining low levels of transient ones. ESQ has variable light conditions (Supplementary Information S1a) influenced by oscillations of the nearby river plume and, recently, has been impacted by iron ore tailings80 which could also have impacted the microbiome richness.
Considering all four host species studied, reef site and host species had equivalent influence on bacterial community structure whereas for the Symbiodiniaceae community hosts were more important than sites. Noteworthy, a gradient of Symbiodiniaceae community structure was seen from widely distributed corals (higher structure) to endemic and specialist hosts (lower structure). Such gradient was also observed in bacterial communities although less pronounced. The much higher taxonomic and functional diversity of bacteria compared to Symbiodiniaceae in association with corals as well as their overspread distribution in coral compartments (mucus and tissues) as opposed to the microalgae (exclusive to the gastrodermis) could explain such weaker gradient in bacteria. The mucus is a more dynamic matrix than the coral tissue, harbors distinct, more variable microbial communities, and may also trap water-borne bacteria on its surface81,82. Differences in the ratio of mucus to tissue among the coral species and across sites for the same species might have influenced the proportion of stable vs. variable bacterial communities.
Depth-generalist corals have more flexible bacteria and Symbiodiniaceae communities than specialist corals18,26. An alternative, not mutually exclusive, explanation for the greater flexibility of the microbial community in response to the environment is that the host population could be spatially structured with a high degree of locally adapted genes, which could be associated with distinct communities19,20. However, as our sampling was conducted across a much smaller area than the aforementioned studies (160 km vs > 7000 km), it is possible that the host genetic variation effect would be less pronounced. Further studies on Abrolhos corals’ genetic diversity are needed to test this hypothesis. Moreover, the coral’s capacity to inhabit variable environments is probably operated by several mechanisms, such as host epigenetic controls83 and phylotypes’ diversity19,84, and functional associations with specific bacterial taxa85. However, bacteria that participate intrinsically in this broad adaptation or benefit sporadically from these corals remain more difficult to determine.
The remaining unexplained variance in the microbial community suggests the influence of stochasticity in community assembly and the influence of interactions within the microbiome. Positive correlations in the well-structured microbial network indicate cooperative interactions while negatively correlated nodes suggest competition for space and resources6,86. Vibrionaceae (mainly Vibrio and Photobacterium) accounted for 49% of the nodes in the main module and was the most abundant family in all F. gravida samples from SG and most samples from TIM. These bacteria are commonly found free-living in coastal waters87,88 and along the Brazilian coast88, where they are widely associated with benthic organisms89. Vibrio includes species with pathogenic potential, efficient quorum-sensing signaling, and a set of mechanisms for efficient infection, competition and resistance90. These copiotrophic strategists might be associated with nitrogen cycling89. Many Vibrio species were associated with dysbiosis and coral bleaching/disease, including white plague in Mussismilia sp. from Abrolhos90,91. The high proportions of vibrios observed in F. gravida could be related to an opportunistic shift in the community. Abnormally high temperatures and low turbidity, with consequently higher irradiance at the bottom in the winter preceding our sampling (Supplementary Information 1b) may have had a cumulative effect on corals. Later, in the austral summer of 2018–2019, the region experienced the most severe thermal anomaly ever registered79. Noteworthy, F. gravida and M. harttii presented the highest bleaching indexes among Abrolhos reef corals during the 2016 thermal anomaly77, which could have been linked to a potential susceptibility to opportunistic vibrios. It has been speculated that hosts harboring flexible microbiomes are more susceptible to opportunistic infections due to unstable associations, especially during transition phases25, an hypothesis that needs more investigation.
The high prevalence and abundance of vibrios in SG corals could also be associated with this site’s high turbidity and sediment deposition from a nearby dredging disposal area88. Relatively high coverage of macroalgae and steadily increasing zoanthid (Palythoa) covers, indicators of anthropogenic impacts, were also reported for this reef33. Additionally, high numbers of vibrios have been recorded in the water column of Abrolhos reefs, mainly in SG and TIM88. Our results indicate an association between environmental factors and host species’ permeability as the main drivers of high Vibrio abundance. Whether the massive abundance of vibrios, especially in F. gravida from SG, was episodic or if it will culminate in a persistent pathobiome is unclear and requires further investigation.
The antagonic cluster to the Vibrionaceae (cluster II) was much more diverse in terms of families than the Vibrionaceae cluster itself (cluster I), markedly in M. harttii and M. cavernosa. Some abundant taxa in cluster II, classified as Sphingomonas and Ralstonia, have been previously observed inside Symbiodiniaceae cells69. Negative interactions, despite being harder to detect, stabilize networks86, meaning clusters I and II stability is largely supported by the antagonism of Vibrio and Sphingomonas.
Conclusions
Our coupled analyses of bacterial and Symbiodiniaceae communities in four coral species from the SAO’s largest and richest coral reef system allowed the exploration of novel multi-taxa associations and interactions. Also, they contributed to revealing novel components of the diversity of microbial communities associated with corals as several novel taxa.
Biological filters governed mainly by the host are essential drivers shaping corals’ microbiome structure. The higher degree of community structure by reef in symbiotic associations appears to be linked with coral species with broader geographic and depth ranges, a feature more prominent in Symbiodiniaceae but also observed to a lesser degree in bacteria. In contrast, endemic and habitat-specialist host, M. braziliensis, seem to has more homogenous bacterial communities with more exclusive members. This lends credence to the hypothesis that higher microbiome flexibility renders corals more adaptable to diverse environments, a trend that should be investigated in more hosts and reef areas.
Data availability
The datasets generated and analysed during the current study are available in the NCBI repository. Data was deposited as Sequence Read Archive (SRA) data with BioProject number PRJNA1012704. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1012704.
References
Rosenberg, E., Koren, O., Reshef, L., Efrony, R. & Zilber-Rosenberg, I. The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5, 355–362 (2007).
Barnes, D. J. Calcification and photosynthesis in reef-building corals and algae. Ecosyst. World 25, 109–131 (1990).
LaJeunesse, T. C. et al. Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr. Biol. 28, 2570–2580 (2018).
Abrego, D., Ulstrup, K. E., Willis, B. L. & van Oppen, M. J. H. Species-specific interactions between algal endosymbionts and coral hosts define their bleaching response to heat and light stress. Proc. Biol. Sci. 275, 2273–2282 (2008).
Cunning, R., Silverstein, R. N. & Baker, A. C. Symbiont shuffling linked to differential photochemical dynamics of Symbiodinium in three Caribbean reef corals. Coral Reefs 37, 145–152 (2018).
Raina, J.-B., Tapiolas, D., Willis, B. L. & Bourne, D. G. Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl. Environ. Microbiol. 75, 3492–3501 (2009).
Lesser, M. P. et al. Nitrogen fixation by symbiotic cyanobacteria provides a source of nitrogen for the scleractinian coral Montastraea cavernosa. Mar. Ecol. Prog. Ser. 346, 143–152 (2007).
Hernandez-Agreda, A., Leggat, W., Bongaerts, P., Herrera, C. & Ainsworth, T. D. Rethinking the coral microbiome: Simplicity exists within a diverse microbial biosphere. MBio 9, 10 (2018).
LaJeunesse, T. C. et al. Long-standing environmental conditions, geographic isolation and host-symbiont specificity influence the relative ecological dominance and genetic diversification of coral endosymbionts in the genus Symbiodinium. J. Biogeogr. 37, 785–800 (2010).
Ziegler, M. et al. Coral bacterial community structure responds to environmental change in a host-specific manner. Nat. Commun. 10, 3092 (2019).
Pootakham, W. et al. Taxonomic profiling of Symbiodiniaceae and bacterial communities associated with Indo-Pacific corals in the Gulf of Thailand using PacBio sequencing of full-length ITS and 16S rRNA genes. Genomics 113, 2717–2729 (2021).
Putnam, H. M., Stat, M., Pochon, X. & Gates, R. D. Endosymbiotic flexibility associates with environmental sensitivity in scleractinian corals. Proc. Biol. Sci. 279, 4352–4361 (2012).
Osman, E. O. et al. Coral microbiome composition along the northern Red Sea suggests high plasticity of bacterial and specificity of endosymbiotic dinoflagellate communities. Microbiome 8, 8 (2020).
Zarate, D., Gary, J. & Li, J. Flexibility in coral-algal symbiosis is positively correlated with the host geographic range. Ecol. Lett. 27, e14374 (2024).
Hume, B. C. C. et al. Symbiodinium thermophilum sp. nov., a thermotolerant symbiotic alga prevalent in corals of the world’s hottest sea, the Persian/Arabian Gulf. Sci. Rep. 5, 8562 (2015).
Meyer, O. Functional groups of microorganisms. In Biodiversity and Ecosystem Function (eds Schulze, E.-D. & Mooney, H. A.) 67–96 (Springer, 1994).
Fabina, N. S., Putnam, H. M., Franklin, E. C., Stat, M. & Gates, R. D. Transmission mode predicts specificity and interaction patterns in coral-Symbiodinium networks. PLoS ONE 7, e44970 (2012).
Bongaerts, P. et al. Prevalent endosymbiont zonation shapes the depth distributions of Scleractinian coral species. R. Soc. Open Sci. 2, 140297 (2015).
Reigel, A. M. & Hellberg, M. E. Microbiome environmental shifts differ between two co-occurring octocoral hosts. Mar. Ecol. Prog. Ser. 720, 59–83 (2023).
Jia, S. et al. Environmental heterogeneity contributes to population genetic diversity and spatial genetic structure of coral-algal symbiosis of Platygyra daedalea in the northern South China Sea. Ecol. Indic. 154, 110599 (2023).
Buzzoni, D., Cunning, R. & Baker, A. C. The role of background algal symbionts as drivers of shuffling to thermotolerant Symbiodiniaceae following bleaching in three Caribbean coral species. Coral Reefs 42, 1285–1295 (2023).
Lawrence, D. et al. Species interactions alter evolutionary responses to a novel environment. PLoS Biol. 10, e1001330 (2012).
Faust, K. & Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 10, 538–550 (2012).
Reshef, L., Koren, O., Loya, Y., Zilber-Rosenberg, I. & Rosenberg, E. The coral probiotic hypothesis. Environ. Microbiol. 8, 2068–2073 (2006).
Voolstra, C. R. & Ziegler, M. Adapting with microbial help: Microbiome flexibility facilitates rapid responses to environmental change. Bioessays 42, e2000004 (2020).
Glasl, B. et al. Microbiome variation in corals with distinct depth distribution ranges across a shallow–mesophotic gradient (15–85 m). Coral Reefs 36, 447–452 (2017).
Ge, R. et al. Regulation of the coral-associated bacteria and Symbiodiniaceae in Acropora valida under ocean acidification. Front. Microbiol. 12, 767174 (2021).
Chen, B. et al. Microbiome community and complexity indicate environmental gradient acclimatisation and potential microbial interaction of endemic coral holobionts in the South China Sea. Sci. Total Environ. 765, 142690 (2021).
Claar, D. C. et al. Increased diversity and concordant shifts in community structure of coral-associated Symbiodiniaceae and bacteria subjected to chronic human disturbance. Mol. Ecol. 29, 2477–2491 (2020).
Quek, Z. B. R. et al. Limited influence of seasonality on coral microbiomes and endosymbionts in an equatorial reef. Ecol. Indic. 146, 109878 (2023).
Lesser, M. P. et al. Photoacclimatization by the coral Montastraea cavernosa in the mesophotic zone: Light, food, and genetics. Ecology 91, 990–1003 (2010).
Moura, R. L. et al. An extensive reef system at the Amazon River mouth. Sci. Adv. 2, e1501252 (2016).
Teixeira, C. D. et al. Decadal (2006–2018) dynamics of Southwestern Atlantic’s largest turbid zone reefs. PLoS ONE 16, e0247111 (2021).
Ribeiro, F. V. et al. Long-term effects of competition and environmental drivers on the growth of the endangered coral Mussismilia braziliensis (Verril, 1867). PeerJ 6, e5419 (2018).
Nunes, F., Sweet, M., Kitahara, M. & Huang, M. V. Mussismilia braziliensis. In IUCN Red List of Threatened Species. https://doi.org/10.2305/iucn.uk.2022-2.rlts.t133586a165962303.en (IUCN, 2021).
Castro, C. B. & Pires, D. O. Brazilian coral reefs: What we already know and what is still missing. Bull. Mar. Sci. 69, 357–371 (2001).
Nunes, F. L. D., Sweet, M., Kitahara, M. V. & Huang, D. Mussismilia harttii. In IUCN Red List of Threatened Species (IUCN, 2021).
Kitahara, M. V., Croquer, A., Alvarez-Filip, L., Banaszak, A. & Nunes, F. Favia gravida. In The IUCN Red List of Threatened Species. https://doi.org/10.2305/iucn.uk.2022-2.rlts.t133237a165787295.en (2021).
Fukami, H. et al. Conventional taxonomy obscures deep divergence between Pacific and Atlantic corals. Nature 427, 832–835 (2004).
Hume, B. C. C. et al. An improved primer set and amplification protocol with increased specificity and sensitivity targeting the Symbiodinium ITS2 region. PeerJ 6, e4816 (2018).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1 (2013).
Hume, B. C. C. et al. SymPortal: A novel analytical framework and platform for coral algal symbiont next-generation sequencing ITS2 profiling. Mol. Ecol. Resour. 19, 1063–1080 (2019).
Arif, C. et al. Assessing Symbiodinium diversity in Scleractinian corals via next-generation sequencing-based genotyping of the ITS2 rDNA region. Mol. Ecol. 23, 4418–4433 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Edgar, R. Usearch (2010).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Team, R. C. & Core Team, R. R: A Language and Environment for Statistical Computing. (R Foundation Statistical Computing).
Oksanen, J. et al. The vegan package. Community Ecol. Pack. 10, 719 (2007).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 (1995).
Ripley, B. et al. Package ‘mass’. Cran R 538, 113–120 (2013).
De Cáceres, M., Jansen, F. & Dell, N. Indicspecies: Relationship between species and groups of sites. R Package Version (2016).
Friedman, J. & Alm, E. J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8, e1002687 (2012).
Weiss, S. et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 10, 1669–1681 (2016).
Watts, S. C., Ritchie, S. C., Inouye, M. & Holt, K. E. FastSpar: Rapid and scalable correlation estimation for compositional data. Bioinformatics 35, 1064–1066 (2019).
Saito, R. et al. A travel guide to Cytoscape plugins. Nat. Methods 9, 1069–1076 (2012).
Assenov, Y., Ramírez, F., Schelhorn, S.-E., Lengauer, T. & Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 24, 282–284 (2008).
Silva-Lima, A. W. et al. Multiple symbiodinium strains are hosted by the Brazilian endemic corals Mussismilia spp.. Microb. Ecol. 70, 301–310 (2015).
Garrido, A. G. et al. Marine heatwave caused differentiated dysbiosis in photosymbiont assemblages of corals and hydrocorals during El Niño 2015/2016. Microb. Ecol. https://doi.org/10.1007/s00248-023-02299-3 (2023).
Teschima, M. M., Garrido, A., Paris, A., Nunes, F. L. D. & Zilberberg, C. Biogeography of the endosymbiotic dinoflagellates (Symbiodiniaceae) community associated with the brooding coral Favia gravida in the Atlantic Ocean. PLoS ONE 14, e0213519 (2019).
van Oppen, M. J. H. & Medina, M. Coral evolutionary responses to microbial symbioses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 375, 20190591 (2020).
Swain, T. D., Lax, S., Gilbert, J., Backman, V. & Marcelino, L. A. A phylogeny-informed analysis of the global coral-symbiodiniaceae interaction network reveals that traits correlated with thermal bleaching are specific to symbiont transmission mode. mSystems 6, 3 (2021).
Mies, M. et al. South Atlantic coral reefs are major global warming refugia and less susceptible to bleaching. Front. Mar. Sci. 7, 514 (2020).
Williams, S. D. & Patterson, M. R. Resistance and robustness of the global coral-symbiont network. Ecology 101, e02990 (2020).
Eckert, R. J., Reaume, A. M., Sturm, A. B., Studivan, M. S. & Voss, J. D. Depth influences Symbiodiniaceae associations among Montastraea cavernosa corals on the belize barrier reef. Front. Microbiol. 11, 518 (2020).
McCauley, M., Goulet, T. L., Jackson, C. R. & Loesgen, S. Systematic review of cnidarian microbiomes reveals insights into the structure, specificity, and fidelity of marine associations. Nat. Commun. 14, 4899 (2023).
Hoffmann, K. et al. Diversity and metabolism of Woeseiales bacteria, global members of marine sediment communities. ISME J. 14, 1042–1056 (2020).
Kaboré, O. D., Godreuil, S. & Drancourt, M. Planctomycetes as host-associated bacteria: A perspective that holds promise for their future isolations, by mimicking their native environmental niches in Clinical Microbiology Laboratories. Front. Cell. Infect. Microbiol. 10, 519301 (2020).
Silva, A. S., Leão, Z. M. A. N., Kikuchi, R. K. P., Costa, A. B. & Souza, J. R. B. Sedimentation in the coastal reefs of Abrolhos over the last decades. Cont. Shelf Res. 70, 159–167 (2013).
Maire, J. et al. Intracellular bacteria are common and taxonomically diverse in cultured and in hospite algal endosymbionts of coral reefs. ISME J. 15, 2028–2042 (2021).
Meunier, V. et al. Bleaching forces coral’s heterotrophy on diazotrophs and Synechococcus. ISME J. 13, 2882–2886 (2019).
Muhammad, S. A. & Ahmed, S. Production and characterization of a new antibacterial peptide obtained from Aeribacillus pallidus SAT4. Biotechnol. Rep. (Amst.) 8, 72–80 (2015).
de Castro, A. P. et al. Bacterial community associated with healthy and diseased reef coral Mussismilia hispida from eastern Brazil. Microb. Ecol. 59, 658–667 (2010).
Paulino, G. V. B. et al. Microbiota of healthy and bleached corals of the species Siderastrea stellata in response to river influx and seasonality in Brazilian northeast. Environ. Sci. Pollut. Res. Int. 30, 26496–26509 (2023).
Silva-Lima, A. W. et al. Mussismilia braziliensis white plague disease is characterized by an affected Coral immune system and dysbiosis. Microb. Ecol. 81, 795–806 (2021).
Lins-de-Barros, M. M. et al. Archaea, bacteria, and algal plastids associated with the reef-building corals Siderastrea stellata and Mussismilia hispida from Búzios, South Atlantic Ocean, Brazil. Microb. Ecol. 59, 523–532 (2010).
Hester, E. R., Barott, K. L., Nulton, J., Vermeij, M. J. & Rohwer, F. L. Stable and sporadic symbiotic communities of coral and algal holobionts. ISME J. 10, 1157–1169 (2016).
Teixeira, C. D. et al. Sustained mass coral bleaching (2016–2017) in Brazilian turbid-zone reefs: Taxonomic, cross-shelf and habitat-related trends. Coral Reefs 38, 801–813 (2019).
Ferreira, L. C. L., Grillo, A. C., Repinaldo Filho, F. P. M., Souza, F. N. R. & Longo, G. O. Different responses of massive and branching corals to a major heatwave at the largest and richest reef complex in South Atlantic. Mar. Biol. 168, 54 (2021).
Corazza, B. M. et al. No coral recovery three years after a major bleaching event in reefs in the Southwestern Atlantic refugium. Mar. Biol. 171, 1 (2024).
Sartori, É. et al. Trace metal concentration along the Brazilian coast: An assessment of the influence of the Doce River plume. Mar. Pollut. Bull. 188, 114640 (2023).
Sweet, M. J., Croquer, A. & Bythell, J. C. Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs 30, 39–52 (2011).
Guppy, R. & Bythell, J. C. Environmental effects on bacterial diversity in the surface mucus layer of the reef coral Montastraea faveolata. Mar. Ecol. Prog. Ser. 328, 133–142 (2006).
Granados-Cifuentes, C., Bellantuono, A. J., Ridgway, T., Hoegh-Guldberg, O. & Rodriguez-Lanetty, M. High natural gene expression variation in the reef-building coral Acropora millepora: Potential for acclimative and adaptive plasticity. BMC Genom. 14, 228 (2013).
Bongaerts, P. et al. Adaptive divergence in a scleractinian coral: Physiological adaptation of Seriatopora hystrix to shallow and deep reef habitats. BMC Evol. Biol. 11, 303 (2011).
Torda, G. et al. Rapid adaptive responses to climate change in corals. Nat. Clim. Change 7, 627–636 (2017).
Coyte, K. Z., Schluter, J. & Foster, K. R. The ecology of the microbiome: Networks, competition, and stability. Science 350, 663–666 (2015).
Alves, N. Jr. et al. Diversity and pathogenic potential of vibrios isolated from Abrolhos Bank corals. Environ. Microbiol. Rep. 2, 90–95 (2010).
Bruce, T. et al. Abrolhos bank reef health evaluated by means of water quality, microbial diversity, benthic cover, and fish biomass data. PLoS ONE 7, e36687 (2012).
Chimetto Tonon, L. A. et al. Diversity and ecological structure of vibrios in benthic and pelagic habitats along a latitudinal gradient in the Southwest Atlantic Ocean. PeerJ 3, e741 (2015).
Zhou, J. et al. Opportunistic bacteria use quorum sensing to disturb coral symbiotic communities and mediate the occurrence of coral bleaching. Environ. Microbiol. 22, 1944–1962 (2020).
Chimetto Tonon, L. A. et al. Quantitative detection of active vibrios associated with white plague disease in Mussismilia braziliensis corals. Front. Microbiol. 8, 2272 (2017).
Acknowledgements
L.B.V thanks Programa de Pós graduação em Genética (UFRJ), FAPERJ, CAPES, and FEST/RENOVA for her doctoral scholarship and resources. The authors are thankful to G.M. Castro, C.D. Teixeira, F.C. Cardoso, H.M. Fortunato, D.S. Cajueiro, I. Pierozzi Jr., A. Shimada, and R.B. Menezes for sampling assistance. L.A. Carlos Jr. (PUC-RJ) and P. Chiroque are acknowledged for their assistance with statistical analyses and J. Gonçalves and T. Louzada for helping with the remote sensing data. P.I. Hargreaves and L.A. Carlos Jr. are acknowledged for their invaluable comments and suggestions. R.L.M. and P.S.S. acknowledge long-term individual grants from CNPq.
Funding
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Fundação Renova, Conselho Nacional de Desenvolvimento Científico e Tecnológico (Grant 310057/2022-1) and PELD Abrolhos (Grant 441418/2020-1).
Author information
Authors and Affiliations
Contributions
LBV, RLM, FVR, AWSL and PSS did the experimental design. LBV, PSS, YA, and APBM wrote the main manuscript text. LBV prepared all figures. APBM contributed with graphs in Figs. 5 and 6. MA made the drawings in Fig. 7. LBV, MEM, APBM did the bioinformatic analysis. LBV prepared the ITS2 libraries and MEM and TT prepared the 16S rRNA gene libraries. LBV and APBM did the statistical analyses. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Villela, L.B., da Silva-Lima, A.W., Moreira, A.P.B. et al. Bacterial and Symbiodiniaceae communities’ variation in corals with distinct traits and geographical distribution. Sci Rep 14, 24319 (2024). https://doi.org/10.1038/s41598-024-70121-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-70121-2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}