
Abstract
Soil salinization, a prevalent form of environmental stress, leads to significant soil desertification and impacts agricultural productivity by altering the internal soil environment, slowing cellular metabolism, and modifying cellular architecture. This results in a marked reduction in both the yield and diversity of crops. Maize, which is particularly susceptible to salt stress, serves as a critical model for studying these effects, making the elucidation of its molecular responses essential for crop improvement strategies. This study focuses on the phytochrome-interacting factor 3 (PIF3), previously known for its role in freezing tolerance, to assess its function in salt stress tolerance. Utilizing two transcript variants of maize ZmPIF3 (ZmPIF3.1 and ZmPIF3.2), we engineered Arabidopsis transgenic lines to overexpress these variants and analyzed their phenotypic, physiological, biochemical, and transcriptomic responses to salt stress. Our findings reveal that these transgenic lines displayed not only enhanced salt tolerance but also improved peroxide decomposition and reduced cellular membrane damage. Transcriptome analysis indicated significant roles of hormonal and Ca2+ signaling pathways, along with key transcription factors, in mediating the enhanced salt stress response. This research underscores a novel role for ZmPIF3 in plant salt stress tolerance, offering potential avenues for breeding salt-resistant crop varieties.
Similar content being viewed by others

Introduction
Light profoundly impacts nearly every aspect of physiological growth in plants, orchestrating a broad spectrum of external environmental signals—including abiotic and biotic stresses—as well as internal signals such as photoperiods and hormones1,2. Plants efficiently utilize light receptors to capture these external cues, which subsequently modulate internal signal levels. This intricate interplay of signals orchestrates various signal transduction pathways that collectively govern plant growth and development3. Central to this regulatory mechanism are the Phytochrome-Interacting Factors (PIFs), which integrate environmental and endogenous signals to direct key developmental processes, including embryo formation. PIFs are increasingly recognized as crucial components in plant biology, with PIF3 being a prominent member involved in phytochrome-mediated signaling processes4. In the absence of light, the inactive phytochrome conformer, Pr, is sequestered in the vacuole, allowing PIFs to accumulate in the nucleus. This accumulation leads to the characteristic yellowing and elongation of seedlings5. Light exposure induces a transformation of Pr to Pfr, which migrates to the nucleus where it interacts with PIF3 to regulate the expression of target genes, thereby initiating photomorphogenesis. PIF3 is particularly noteworthy as the first bHLH protein within the PIF family known to interact with both phyA and phyB, affecting the promoter regions of key circadian elements such as CCA1 and LHY, and modulating their expression6,7.
Alternative splicing (AS) is a prevalent mechanism within the plant kingdom that significantly influences various processes including growth, development, signaling transport, circadian rhythms, and responses to abiotic stress8. In Arabidopsis, a significant proportion of intron-retaining genes demonstrate variable splicing patterns that contribute to adaptative responses9. Notably, AS in genes associated with the Arabidopsis thaliana (A. thaliana) circadian clock also plays a role in mediating defense responses10,11. For instance, the gene CCA1 produces two protein variants via AS, which under low-temperature conditions, shifts in splicing patterns enhance frost resistance12,13.
Soil salinization poses a severe challenge by disrupting the internal soil environment through elevated Na+ ion concentrations, decelerating cellular metabolism, and deteriorating cellular structures, significantly impacting crop yield and quality14,15. Maize, a salt-sensitive crop, exhibits limited tolerance and under salt stress conditions, shows reduced growth and productivity16,17. Thus, unraveling the molecular mechanisms of salt stress response is critical for advancing maize breeding strategies and enhancing salt tolerance. The maize PIF family has been extensively studied for over a decade, with research elucidating their roles in light signaling, photomorphogenesis, and abiotic stress tolerance, particularly in drought and salt stress conditions18,19,20,21,22. Among these, ZmPIF3 has been the subject of detailed functional studies, including overexpression analysis in rice as a host plant18. Recently, PIF3 was identified as a negative regulator of salt tolerance in Arabidopsis, with its stability being compromised by SOS2-mediated phosphorylation23. These findings highlight the complex and multifaceted roles of PIF3 in plant stress responses and development.
This study employs two alternative splicing variants of maize ZmPIF3, namely ZmPIF3.1 and ZmPIF3.2, to generate overexpressing transgenic lines of A. thaliana. We evaluate their phenotypic, physiological, and biochemical responses under varying salt stress conditions and perform a comprehensive transcriptomic analysis. Our aim is to elucidate the regulatory networks and molecular mechanisms through which ZmPIF3 contributes to enhanced salt tolerance, potentially paving the way for the development of robust salt-tolerant crops.
Materials and methods
Plant materials
The maize inbred line CML288, sourced from the International Maize and Wheat Improvement Center (CIMMYT) in Mexico, was used for this study. A. thaliana experiments involved the wild-type (WT) ecotype Columbia-0 (Col-0). All Arabidopsis seeds underwent surface sterilization before being germinated on half-strength Murashige and Skoog (MS) medium. The seedlings were then transplanted into sandy soil and grown in a greenhouse under long day (LD) conditions, with temperatures set at 25 °C during a 16-h light period and 23 °C during an 8-h dark period.
Cloning of ZmPIF3 cDNA and bioinformatics analysis
Total RNA was extracted from the leaves of five-leaf stage seedlings of CML288 under short-day conditions using TRIzol reagent (Invitrogen, CA, USA). First-strand cDNA synthesis was performed using the PrimeScript™ 1st Strand cDNA Synthesis Kit (Takara, Tianjin, China). The ZmPIF3 gene was amplified using primers designed from its cDNA sequence (MaizeGDB Gene ID: Zm00001d024783) using Primer Premier 6 software. Sequence analysis and alignment were conducted with DNAMAN version 9.0 (Lynnon Biosoft, Quebec, Canada). The open reading frame (ORF) of ZmPIF3 was identified using the ORF Finder Tool (NCBI). Further bioinformatics analysis, including molecular weight and isoelectric point (pI) predictions, were performed using the Expasy server, with domain prediction via the Conserved Domain Search Service (CD Search) and SMART software.
Transformation of two splicing variants of ZmPIF3 into A. thaliana
The entire open reading frames (ORFs) of two splicing variants, ZmPIF3.1 and ZmPIF3.2, were cloned into the pCAMBIA1304 vector under the control of the CaMV 35S promoter. These constructs were then transformed into WT A. thaliana using the Agrobacterium tumefaciens-mediated floral dip method24. The plants expressing the empty transformation vector as a Control line. To establish homozygous lines, transformed plants were selected with hygromycin at a concentration of 35 µg/ml, and segregation ratios were carefully analyzed. Primers for these constructs were designed using Primer Premier 6 software (Table S1). For salt stress experiments, seeds from T4 homozygous transgenic lines and WT Arabidopsis were used.
Salt stress treatment of transgenic A. thaliana
To assess salt stress tolerance during germination, surface-sterilized seeds (fifty seeds per plate, with three replicates) were placed on half-strength Murashige and Skoog (MS) medium supplemented with 0, 100, 120, or 140 mmol/L NaCl. The germination rate (defined as radicle emergence) and primary root length of both WT and transgenic Arabidopsis seeds were recorded over 10 days under LD conditions for each plate. The Student’s t-test was employed to evaluate differences between WT and transgenic plants.
During post-germination stages, salt stress assays were conducted on transgenic Arabidopsis seedlings. Transgenic plants were initially grown on plates containing 35 mg/L hygromycin, whereas WT seeds were cultivated on standard half-strength MS medium. Four days post-germination, both WT and transgenic plants were transferred to the same soil mixture in the greenhouse as previously described. After three weeks, watering was withheld before the seedlings were irrigated from the bottom of the pots with a 350 mM NaCl solution until the soil was fully saturated. Phenotypic differences between transgenic plants and control groups were monitored until the NaCl solution was removed, and normal cultivation was resumed.
Physiological analysis
To evaluate the physiological differences between WT and transgenic A. thaliana in response to salt stress, 25-day-old seedlings were irrigated with a NaCl solution as described previously. Overground leaves were harvested 4- and 7-days post-treatment for the analysis of malondialdehyde (MDA), proline, and catalase (CAT) activity. Samples collected prior to salt stress served as controls. The MDA content and CAT activity measurements were conducted following the methods outlined by Lv et al.25. The determination of free proline content was performed according to Bates et al.26.
RNA-seq analysis of transgenic A. thaliana plants under salt stress
For the RNA-seq analysis, both WT and transgenic A. thaliana plants were collected after four and seven days of salt stress treatment. Ten whole seedlings, including the overground parts, were pooled for each replicate, with two replicates conducted for the study. All samples were immediately flash-frozen in liquid nitrogen and subsequently stored at − 80 °C. Total RNA extraction was performed using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), followed by DNase I treatment and purification with magnetic oligo (dT) beads.
The RNA samples underwent 150-bp paired-end sequencing on the Illumina HiSeq 2500 platform (Illumina) at the Beijing Genomics Institute, Beijing, China. High-quality reads were obtained by trimming raw sequences using Trimmomatic and FastQC. These reads were then mapped to the TAIR10 A. thaliana reference genome using TopHat software27. Expression level changes and associated q-values (false discovery rate adjusted P-values) were calculated using Cufflinks (version 2.0.2) and Cuffdiff28. Genes were classified as significantly differentially expressed if they met the following criteria: q ≤ 0.05 and |fold change| ≥ 2.
Functional and pathway annotations for the genes were performed using the Gene Ontology (GO) database (http://www.geneontology.org/) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.kegg.jp). Principal component analysis and volcano plots were generated using OmicShare tools, a comprehensive online platform for data analysis (http://www.omicshare.com/tools), to compare all identified genes across the eight samples.
Gene expression quantification by quantitative real-time RT-PCR
Total RNA was extracted from leaves of the maize inbred line CML288, using the same batch and procedure as described for the RNA-seq samples, with TRIzol reagent (Invitrogen, CA, USA). First-strand cDNA synthesis was conducted using the HiScript QRT SuperMix for qPCR with gDNA Eraser (Vazyme, Nanjing, China). Quantitative real-time RT-PCR (qRT-PCR) assays were performed on a 7500 Real-Time PCR System using the SYBR Green I kit (Invitrogen, CA, USA). Primers were designed with Primer Premier 6 software. The relative expression levels of the target genes were analyzed using the 2−ΔΔCt method29. The A. thaliana ACTIN gene served as the endogenous reference. All procedures were replicated three times to ensure data reliability.
Plants materials
The authors confirm that the use of plants in the present study complies with international, national and institutional guidelines.
Statistical analysis
Statistical analysis was performed using GraphPad Prism V.8.0.2. Data were presented as means ± standard deviation (SD). Statistical analysis of all experimental data was completed by ANOVA followed by a post-hoc test. P < 0.05 was considered statistically significant.
Results
Cloning and sequence analysis of ZmPIF3
Utilizing homologous cloning techniques, we successfully amplified two distinct PCR products corresponding to the splicing variants ZmPIF3.1 and ZmPIF3.2 from maize inbred line CML288. Subsequent sequencing analysis confirmed the presence of these variants, which differ primarily in their coding sequences, potentially affecting their function under stress conditions. ZmPIF3.1 was characterized by an open reading frame (ORF) of 1599 base pairs, encoding a protein of 533 amino acids with an isoelectric point (pI) of 6.67. Conversely, ZmPIF3.2 displayed a slightly longer ORF at 1623 base pairs, coding for a protein of 541 amino acids.
Multiple sequence alignment highlighted regions of high conservation interspersed with specific divergent amino acids that suggest functional specificity related to stress response. These alignments were crucial for identifying domains responsible for differential responses to environmental stresses, including salt stress. The sequence analysis, conducted with DNAMAN software, provided insights into potential structural and functional variations between the splice variants (Fig. 1). The alignment of the two ZmPIF3 protein variants with AtPIF3 revealed significant differences, with AtPIF3 showing considerable sequence divergence and low similarity (Fig. S1). These sequence and bioinformatics analyses provide a detailed understanding of the structural variations and potential functional implications of the ZmPIF3 splice variants. These data are essential for elucidating how these variants may influence plant responses to abiotic stresses, particularly salt stress.

Comparative Sequence Alignment of ZmPIF3 Protein Variants. This figure presents a multiple sequence alignment of the ZmPIF3.1 and ZmPIF3.2 splice variants. Identical amino acids between the two sequences are indicated by asterisks (*) directly beneath the aligned residues. Gaps, denoted by dashes (-), have been introduced to maximize the alignment and are indicative of sequence divergence or regions of alternative splicing. The alignment begins from the N-terminal (left) to the C-terminal end (right) of the protein sequences. The residue positions are numbered sequentially from the N-terminus for each variant along the right margin. The highlighted differences in the sequence alignment could suggest functional divergence between the splice variants, potentially leading to variations in their regulatory roles under salt stress conditions.
Overexpression of ZmPIF3.1 in A. thaliana enhances salt stress tolerance
To investigate the role of ZmPIF3 in salt stress response, we constructed overexpression vectors for its two splicing variants, ZmPIF3.1 and ZmPIF3.2, and introduced them into A. thaliana. The impact of these modifications was assessed by examining PIF3 expression levels in the overexpressed transgenic seedlings (OEs) through quantitative real-time PCR (qRT-PCR) analysis (Fig. 2). This assessment revealed a significant increase in PIF3 expression in the transgenic lines compared to the WT and Control lines.

Comparative Analysis of ZmPIF3 Expression in Transgenic and WT A. thaliana. This bar graph depicts the relative expression levels of the ZmPIF3 gene in transgenic A. thaliana overexpressing either ZmPIF3.1 (ZmPIF3.1-OE1; ZmPIF3.1-OE2) or ZmPIF3.2 (ZmPIF3.2-OE1; ZmPIF3.2-OE2) compared to the WT and Control plants expressing the empty transformation vector. Expression levels are normalized to the WT, which is set to a value of 1. Statistical significance is denoted by asterisks, with *** indicating p < 0.001. Data are presented as mean ± SEM.
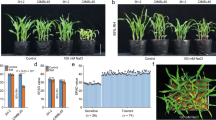
Phenotypic observations between the WT and overexpressed lines under normal conditions (Normal) showed no significant differences in root traits. However, under salt concentrations (100 mmol L−1, 120 mmol L−1 and 140 mmol L−1 NaCl), distinct differences emerged. Notably, the germination rate and root length of the ZmPIF3.1 transgenic lines significantly exceeded those of Control, WT and ZmPIF3.2 transgenic plants (Fig. 3a). Specifically, the root length of ZmPIF3.1 transgenic plants was nearly double that of the WT and ZmPIF3.2 transgenic lines at 140 mmol L−1 NaCl, and there was an approximate 20% increase in germination rate in ZmPIF3.1 overexpressed lines compared to the other two lines (Fig. 3b). These results demonstrate that overexpression of ZmPIF3.1 leads to enhanced salt tolerance compared to ZmPIF3.2 overexpression.

Salt Stress Response in Transgenic and WT Arabidopsis thaliana Seedlings. (a) This bar chart represents the relative root length of transgenic Arabidopsis overexpressing ZmPIF3.1 (ZmPIF3.1-OE1; ZmPIF3.1-OE2) or ZmPIF3.2 (ZmPIF3.2-OE1; ZmPIF3.2-OE2) versus WT and Control plants under different salt concentrations (Normal: control condition, 100 mmol/L, 120 mmol/L, and 140 mmol/L NaCl). Root length is measured in centimeters (cm). Under control conditions, there is no significant difference in root length between the genotypes. However, at elevated salt concentrations, ZmPIF3.1-OE exhibits notably longer roots compared to both WT and ZmPIF3.2-OE seedlings. (b) This bar chart displays the relative germination rate of the same plant groups under the same salt stress conditions. Germination rates are shown as a percentage (%) of the total number of seeds. Similar to root length, ZmPIF3.1-OE seeds demonstrate a higher germination rate under salt stress conditions, suggesting increased salt tolerance. In both panels, lowercase letters indicate statistically significant differences between groups under the same treatment. Error bars represent the standard error of the mean (SEM).
Further validation was sought to determine if overexpression of ZmPIF3.1 could improve salt tolerance during the post-germination phase. Germinated transgenic seeds and vigorous WT seeds were transplanted onto half-strength MS medium containing 350 mmol L−1 NaCl. After four days, a clear distinction was observed: while WT plants showed a decline in salt tolerance, transgenic A. thaliana plants thrived, exhibiting lush, extended leaves (Fig. 4). OE plants also had a higher survival rate after salt treatment (Fig. S2). These findings provide strong evidence that overexpression of ZmPIF3.1 significantly enhances the salt stress tolerance of A. thaliana.

Phenotypic Response of ZmPIF3.1-Overexpressing Transgenic Arabidopsis and WT Plants to Salt Stress. This figure displays a side-by-side comparison of ZmPIF3.1-overexpressing transgenic Arabidopsis thaliana (OE1, OE2), WT, and Control plants under conditions of salt stress (350 mmol/L NaCl) and Normal conditions. The top panel shows the seedlings 4 days after treatment (4 d), while the bottom panel shows the seedlings 7 days after treatment (7 d). The transgenic plants exhibit a visibly healthier phenotype under salt stress compared to the WT, as evidenced by a greener color and more robust growth. Under normal conditions, both transgenic and WT plants appear similarly healthy. These images visually demonstrate the enhanced salt tolerance conferred by the overexpression of the ZmPIF3.1 gene in Arabidopsis thaliana.
Physiological changes in ZmPIF3.1 overexpressing A. thaliana under salt stress
Physiological analyses under normal conditions revealed no significant differences between WT and transgenic A. thaliana plants. However, after four days of exposure to 350 mM NaCl, all lines showed elevated levels of malondialdehyde (MDA), proline, and catalase (CAT) activity (Fig. 5). Following salt stress, ZmPIF3.1 overexpressing plants exhibited a substantial increase in proline content, significantly surpassing that of untreated plants and markedly exceeding levels in WT plants (Fig. 5a). This indicates that overexpression of ZmPIF3.1 enhances proline synthesis, thereby potentially increasing drought stress resilience. Moreover, CAT activity was measured, revealing higher accumulation in transgenic plants, suggesting that overexpression of ZmPIF3.1 may mitigate reactive oxygen species (ROS) accumulation by enhancing CAT activity under salt stress. In contrast, MDA content in the transgenic lines was lower than in WT plants, with reductions of 1.30 and 1.33 times after four and seven days of salt stress, respectively (Fig. 5b,c). These results highlight the potential of overexpressing ZmPIF3.1 in A. thaliana to induce significant physiological adaptations conducive to salt stress resilience.

Physiological and Biochemical Responses of Transgenic and WT Arabidopsis to Salt Stress. (a) Proline content (µg/g FW) in transgenic and WT Arabidopsis thaliana seedlings measured at 0, 4, and 7 days post salt stress treatment. C1 and C2 represent WT seedlings under normal and salt stress conditions, respectively, while T1 and T2 represent ZmPIF3.1-overexpressing transgenic seedlings under the same conditions. Proline content increases in response to salt stress, with T2 showing the highest levels, indicating an enhanced stress response. (b) Catalase (CAT) content (µmol/g) under the same conditions and time points. Similar to proline, CAT content increases in response to salt stress, with transgenic seedlings (T2) exhibiting higher CAT levels than WT (C2), suggesting improved reactive oxygen species scavenging capability. (c) Malondialdehyde (MDA) levels (µmol/g FW), a marker of lipid peroxidation, indicating cellular damage. While MDA levels increase under salt stress for all plants, transgenic seedlings (T2) show lower MDA content compared to WT under stress (C2), pointing to reduced oxidative damage in transgenic lines. In all panels, the lowercase letters above the bars denote significant differences at p < 0.05 as determined by ANOVA followed by a post-hoc test. Error bars represent standard error of the mean (SEM).
Genome-wide gene expression changes in ZmPIF3.1 overexpressing plants in response to salt stress treatment
To investigate the transcriptional changes induced by the overexpression of ZmPIF3.1 in response to salt stress, we conducted a comparative transcriptomic analysis between ZmPIF3.1 overexpressing A. thaliana and WT plants under both normal and salt stress conditions. After filtering out low-quality reads, the percentage of mapped unique reads for each sample exceeded 84% (Table 1). Principal Component Analysis (PCA) was utilized to verify the reliability of the transcriptome data across different biological replicates. The first two principal components, PC1 and PC2, accounted for 63.8% and 10.9% of the variation, respectively. Under both normal and salt stress conditions, the replicate distributions for both plant types were closely clustered (Fig. 6a), indicating a robust experimental design.

Transcriptome Analysis of Transgenic and WT Arabidopsis under Salt Stress. (a) Principal Component Analysis (PCA) demonstrating the variance in gene expression between WT and transgenic Arabidopsis under normal (C1, C2) and salt stress (T1, T2) conditions. The PCA plot shows the clustering of biological replicates, with PC1 and PC2 explaining 63.8% and 10.9% of the variance, respectively. (b,c) Volcano plots displaying the differentially expressed genes between WT and transgenic Arabidopsis under normal (b) and salt stress conditions (c). Genes with log2 fold change (FC) and statistical significance (-log10 p-value) are plotted, showing up-regulated (red) and down-regulated (green) genes. (d) Venn diagram illustrating the overlap of differentially expressed genes between WT and transgenic Arabidopsis under salt stress conditions, highlighting shared and unique gene expression changes. (e) Bar graph depicting the total number of differentially expressed genes, including the count of up-regulated and down-regulated genes in WT and transgenic Arabidopsis under salt stress conditions.
In total, 6152 differentially expressed genes (DEGs) were identified in the transgenic Arabidopsis, compared to 3983 DEGs in the WT (Fig. 6b–d). Of these, 3167 were up-regulated and 2985 were down-regulated in the transgenic plants in response to salt stress, while in the WT, 2056 DEGs were up-regulated and 1927 were down-regulated (Fig. 6e). Notably, approximately 3707 DEGs were common between the two groups, with an additional 276 genes differentially expressed exclusively in the WT plants, and 2445 genes showing unique differential expression in the transgenic plants (Fig. 6d).
To further understand the functional implications of these DEGs, we analyzed them using the Gene Ontology (GO) database. The DEGs associated with cellular components were notably enriched in three main GO categories: biological processes, cellular components, and molecular functions. Key terms such as "cells", "cell parts", "membranes", and "organelles" were prominent under the "cellular component" category. The "molecular function" category included terms like "binding" and "catalytic activity", while "metabolic processes" and "cellular processes" were prevalent in the biological processes category (Fig. 7a). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that these genes were primarily involved in "metabolic pathways", followed by "biosynthesis of secondary metabolites", "biosynthesis of antibiotics", "plant hormone signal transduction", "carbon metabolism", and "biosynthesis of amino acids" (Fig. 7b). This comprehensive analysis underscores the critical roles these DEGs play in the salt stress response mediated by the overexpression of ZmPIF3.1.

Functional Analysis of Differentially Expressed Genes (DEGs) in ZmPIF3 Transgenic and WT Arabidopsis. (a) Gene Ontology (GO) enrichment analysis of DEGs. The bar chart illustrates the percentage of genes within various GO categories, comparing ZmPIF3 transgenic A. thaliana (blue) and WT (green) plants. The y-axis represents the percent of genes, and the x-axis lists the GO terms related to biological processes, cellular components, and molecular functions. Enriched GO terms are evident in several biological processes, particularly in ‘cellular processes’ and ‘metabolic processes’, indicating active reprogramming of these pathways in response to transgenic expression of ZmPIF3. (b) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway categories of DEGs. This histogram displays the distribution of DEGs across different KEGG pathways for ZmPIF3 transgenic (blue) and WT (red) plants. The number of genes assigned to each pathway category is displayed on the y-axis, while the KEGG pathway names are on the x-axis. The transgenic lines show a pronounced enrichment in pathways such as ‘carbohydrate metabolism’, highlighting alterations in metabolic pathways due to overexpression of ZmPIF3.
Analysis of top differentially expressed genes associated with salt tolerance
We categorized all differentially expressed genes (DEGs) into three profiles: specifically expressed in the WT (I); specifically expressed in transgenic plants (II); and differentially expressed in both WT and transgenic plants (III). To explore their roles, we performed functional annotations for 50 significantly differentially expressed genes within each profile. The results revealed that a majority of these genes are intricately linked to hormone regulation, calcium regulation, and transcription factor regulation.
In total, 32 DEGs were involved in hormone regulation, predominantly associated with the regulation of ethylene (10/32, 31%), auxin (11/32, 34%), and abscisic acid (3/32, 9%) (Table 2). Additionally, 10 DEGs related to the regulation of calcium metabolic mechanisms were primarily found in profiles II and III, with 70% of these genes showing up-regulated expression (Table 2). These findings suggest that ZmPIF3.1 may enhance salt tolerance by regulating these genes.
Furthermore, 15 transcription factors were differentially expressed under salt stress, including MYB, ERF, bHLH, CBF, bZIP, and WRKY transcription factor families. Among these, MYB, ERF, and CBF transcription factors were relatively predominant, accounting for 27%, 27%, and 20% of the transcription factors, respectively. MYB transcription factors were present in all three profiles, and ERF transcription factors were found in profiles II and III, both predominantly up-regulated. However, CBF transcription factors were only identified in profile II and were also up-regulated (Table 2), indicating their potential roles in enhancing salt tolerance. Collectively, these results suggest that ZmPIF3.1 may influence salt stress response through the regulation of key genes involved in hormone and calcium signaling, as well as through the modulation of critical transcription factors.
Validation of differentially expressed genes using RT-qPCR
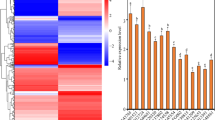
To further verify the reliability of our transcriptome data, we randomly selected 10 differentially expressed genes for quantitative analysis using real-time quantitative PCR (RT-qPCR). These genes included SOS3-interacting protein 3 (SIP3; AT4G30960), which encodes CBL-interacting protein kinase 6 (CIPK6); trehalose-6-phosphate phosphatase G (TPPG; AT4G22590), a gene for a haloacid dehalogenase-like hydrolase (HAD) superfamily protein; MYB domain protein 60 (MYB60; AT1G08810), a putative transcription factor of the R2R3-MYB gene family; Early Arabidopsis aluminum induced 1 (EARLI1; AT4G12480), coding for a putative lipid transfer protein; and NAC domain containing protein 80 (NAC080; AT5G07680), which produces a NAC domain-containing protein. Additionally, ethylene responsive element binding factor 5 (ERF5; AT5G47230) encodes a member of the ERF/AP2 transcription factor family; MYB domain protein 112 (MYB112; AT1G48000) encodes a putative transcription factor; nicotianamine synthase 4 (NAS4; AT1G56430) codes for a protein with nicotianamine synthase activity; beta glucosidase 18 (BGLU18; AT1G52400) is responsible for a member of the glycosyl hydrolase family; and NAC domain containing protein 42 (NAC042; AT2G43000) encodes a NAC transcription factor induced by hydrogen peroxide. The RT-qPCR results demonstrated that the expression profiles of these 10 genes were consistent with those observed in our RNA-Seq data, exhibiting a correlation coefficient (R2) of greater than 0.990 (Fig. 8). This high degree of correlation further substantiates the accuracy of the RNA-Seq data.

Validation of Gene Expression Data by qRT-PCR and RNA-Seq. (a) Correlation analysis of gene expression ratios obtained by qRT-PCR and RNA-Seq for WT Arabidopsis under salt stress (T1) relative to normal conditions (C1). (b) Correlation analysis for ZmPIF3.1-overexpressing transgenic Arabidopsis (T2) relative to normal conditions (C2). Both panels display a scatter plot where each point represents a selected gene, plotting the log2-transformed expression ratio by RNA-Seq on the y-axis against the log2-transformed expression ratio by qRT-PCR on the x-axis. The solid line represents the linear regression fit, and the correlation coefficient (R2) indicates a high degree of correlation between the two methods for both WT and transgenic Arabidopsis, validating the RNA-Seq data.
In summary, our research successfully identified and characterized two splice variants of ZmPIF3, ZmPIF3.1 and ZmPIF3.2, from maize, and evaluated their effects on salt stress tolerance in transgenic A. thaliana. Through detailed cloning, sequence analysis, and bioinformatics, we elucidated distinct structural features that potentially influence their stress response functions. Overexpression of ZmPIF3.1 significantly enhanced salt tolerance, demonstrated by improved germination rates and root growth under saline conditions. Our physiological and transcriptomic analyses revealed substantial changes in biochemical markers and gene expression profiles, suggesting that ZmPIF3.1 alters essential metabolic and stress response pathways. Validation of differentially expressed genes through RT-qPCR corroborated our RNA-seq data, further confirming the role of ZmPIF3.1 in enhancing plant resilience to salt stress.
Discussion
Role of the active oxygen scavenging system in regulating salt tolerance mediated by ZmPIF3.1
The content of malondialdehyde (MDA) in plants is an indicator of membrane peroxidation, providing an indirect measure of damage to the endomembrane system and overall plant stress resistance30. Similarly, the accumulation of proline is generally higher in drought-tolerant varieties and serves as a reflective indicator of stress resistance, which can be assessed by measuring proline levels31. In transgenic A. thaliana characterized by increased free proline accumulation, we observed enhanced salt tolerance. This suggests that organisms with high reactive oxygen species (ROS) scavenging capacity typically exhibit strong stress resistance32, indicating that ZmPIF3.1 transgenic Arabidopsis can decompose peroxide more effectively than the WT, resulting in lower membrane damage and improved salt tolerance.
Osmotic disorders and persistent salt stress can lead to cellular water loss, culminating in reduced cell turgor. To counteract this, plants enhance their cell osmotic potential through the accumulation of metabolites such as proline, sugars, and sugar alcohols, which help reduce the cytoplasmic osmotic potential at relatively high concentrations, thereby preventing the inhibition of cell metabolism33,34. In the plant realm, numerous proteins contribute to this defense mechanism by scavenging ROS. Plants respond to ROS through both active and non-enzymatic scavenging mechanisms, including compounds like ascorbic acid, glutathione, and phenolic compounds. Enzyme-mediated ROS scavenging includes superoxide dismutase, catalase, and glutathione peroxidase35,36. Notably, the capacity of active oxygen scavenging mechanisms correlates with salt tolerance35,37, as demonstrated by transgenic Arabidopsis with enhanced reactive oxygen scavenging capabilities, which showed improved salt tolerance in this study.
Our current investigation revealed significant increases in proline content and catalase activity in ZmPIF3.1 overexpressing transgenic Arabidopsis under salt stress, compared to WT plants. Furthermore, the levels of salt-induced stress were substantially reduced in transgenic Arabidopsis when contrasted with their WT counterparts. These findings underscore the significance of osmotic adjustment substances and the effective elimination of reactive oxygen species as pivotal contributors to enhancing plant salt tolerance.
Regulatory impact of ZmPIF3.1 overexpression on hormone-related genes in salt tolerance
Research has highlighted the critical role of plant hormones such as abscisic acid (ABA), ethylene, and auxin in responding to salt stress38. Salt-induced stress often triggers shifts in the expression of genes associated with auxin and related pathways39. ABA plays a pivotal role in enhancing the survival of seedlings under such conditions40,41. In this study, we observed that the ABA-associated CYP707A3 gene was up-regulated in transgenic A. thaliana, consistent with previous findings of increased CYP707A3 expression under various stress conditions42. In WT plants, the expression level of the CYP707A3 gene post-salt treatment was lower compared to the control, whereas in transgenic lines, it was significantly higher than that of the control. This suggests that ZmPIF3.1 may enhance the expression of this gene to improve salt tolerance.
Additionally, the HOS3-1 gene, known to inhibit ABA-mediated stress responses43, and the SHY2/IAA3 gene, a negative regulator of ethylene signaling that inhibits root elongation44, were examined. The SHY2 gene was down-regulated in both transgenic and WT A. thaliana, with a more pronounced down-regulation observed in the transgenic lines, correlating with the salt-resistant phenotype of transgenic Arabidopsis. This suggests that overexpression of ZmPIF3.1 may directly or indirectly regulate the SHY2 gene, contributing to root elongation in transgenic lines.
These observations imply that ZmPIF3.1 likely modulates salt stress response by regulating the expression of these hormone-related genes, further underscoring the significant role of hormone pathways in plant salt tolerance research.
Overexpression of ZmPIF3.1 regulates calcium-related genes to improve salt tolerance
Calcium ions are crucial second messengers in various environmental stress responses, including salt stress45. Key components of this signaling network are calcium signal receptors such as calcineurin B-like proteins (CBLs) and calmodulins (CAMs)46. In our study, we observed significant alterations in the expression profiles of calcium-related genes, including CMLs and CBLs, under salt stress, highlighting their critical role in mediating salt stress signals.
Previous research has shown that the BnCBL1 gene plays a role in responding to salt stress and phosphate deficiency47. In our experiments, this gene was significantly up-regulated under stress conditions, which correlated with enhanced salt tolerance and increased resilience to low phosphate levels. The CBL1 gene was also up-regulated in transgenic A. thaliana, indicating that overexpression of ZmPIF3.1 may boost CBL1 expression, thereby enhancing the plant's ability to manage salt stress.
These findings suggest that ZmPIF3.1 contributes to the regulation of salt damage by modulating the expression of calcium-related genes. This underscores the importance of calcium-signaling-related genes in research on plant salt tolerance.
Overexpression of ZmPIF3.1 regulates transcription factor-related genes to enhance salt stress response
Understanding the mechanisms that underlie plant stress resistance is pivotal for improving crop yield and resilience. Recent research highlights the role of the circadian clock in regulating genes associated with freeze tolerance, such as the C-repeat binding transcription factor (CBF) gene and its target gene expression, which exhibit a photoperiod-dependent pattern48,49. The induction of freeze tolerance via the CBF gene is also time-dependent50. Direct evidence supports the involvement of circadian clock genes in enhancing low-temperature tolerance, with the CCA1 transcription factor directly binding to the CBF gene promoter, modulating its expression and conferring freezing tolerance51.
In this study, the CBF1, CBF2, and CBF3 genes showed up-regulation under salt stress conditions in transgenic A. thaliana, with a greater extent of up-regulation compared to that in WT Arabidopsis. All CBF genes in leaf tissue were up-regulated in response to salt stress. Studies have indicated that PIF3 can act as a negative regulator of antifreeze mechanisms by binding to the CBF promoter region and inhibiting their expression52. We conclude that ZmPIF3.1 can directly and indirectly interact with CBF to positively modulate CBF gene expression, thereby enhancing salt tolerance in transgenic A. thaliana. Additionally, our findings suggest that ZmPIF3.1 may orchestrate the expression of salt-tolerant genes through the regulation of transcription factors via calcium signaling pathways, or modulate the salt stress response through hormonal pathways. Drawing from our analytical findings and previous research, we infer that ZmPIF3.1 participates in a straightforward pathway for salt regulation, paving the way for deeper exploration into the molecular mechanisms and regulatory networks that govern ZmPIF3.1's response to salt stress.
Role of alternative splicing of the ZmPIF3 gene in salt stress regulation
Alternative splicing is a pivotal molecular mechanism in plants that influences the functionality of the circadian clock26. Many circadian genes show variability in their splicing patterns in response to environmental changes, highlighting the interplay between circadian clock splicing diversity and environmental responsiveness12. Notably, the splicing variability of the CCA1 gene has been linked to freeze-tolerance regulation, producing two protein isoforms through alternative cleavage: CCA1α and CCA1β13. Under normal conditions, CCA1β, which lacks the MYB DNA domain, forms a non-functional heterodimer with full-length CCA1α, suppressing CCA1α's activity. In contrast, cold temperatures reduce the splicing diversity of CCA1, increasing the content of CCA1β protein and enhancing the activity of CCA1α, thereby improving the frost resistance of A. thaliana33. This connection illustrates how the alternative splicing of circadian genes is linked to temperature responses. In our current study, we observed that different transcript variants of ZmPIF3 in transgenic A. thaliana exhibit varying levels of tolerance to salt stress, indicating that the alternative splicing of ZmPIF3 may play a crucial role in the plant's response to salt stress. While our study provides valuable insights into the role of ZmPIF3 in regulating salt tolerance in maize, we acknowledge the importance of exploring additional novel aspects to further enhance the understanding and significance of our research. Our study utilized two independent transgenic lines and an empty vector control line, though ideally, three or more lines would better rule out background effects. We acknowledge this limitation and believe that including additional lines is necessary for more robust conclusions. One promising area for future investigation is the occurrence of alternative splicing of the ZmPIF3 gene in natural populations. Alternative splicing is a common mechanism that allows a single gene to produce multiple protein isoforms, potentially leading to functional diversity. Investigating the alternative splicing patterns of ZmPIF3 across different maize genotypes and environmental conditions could uncover new regulatory mechanisms that contribute to salt tolerance and other stress responses.
Another intriguing aspect is the function of the short N-terminal peptide in the ZmPIF3.2 isoform. Preliminary data suggest that this peptide might play a crucial role in modulating the stability, localization, or interaction of ZmPIF3.2 with other proteins or signaling pathways. Detailed functional analysis of this N-terminal region, including its potential post-translational modifications and interacting partners, could provide deeper insights into the molecular mechanisms underlying ZmPIF3-mediated stress responses.
Differential roles of ZmPIF3 and AtPIF3 in regulating salt tolerance: species-specific mechanisms and environmental interactions
The comparative sequence alignment between ZmPIF3 variants and AtPIF3 indicates substantial divergence, suggesting that the functional roles of PIF3 in maize and Arabidopsis may differ significantly. The specific divergent amino acids in AtPIF3 might confer unique regulatory properties, potentially explaining the observed differences in stress response mechanisms between these species. These findings highlight the importance of species-specific adaptations and suggest that the functional specificity of PIF3 proteins could be critical for optimizing stress tolerance strategies in different plant species.
Our study highlights the complex and multifaceted role of PIFs in plant responses to salt stress. Specifically, we observed that ZmPIF3 modulates salt tolerance in maize under long day photoperiod conditions (16 h light/8 h dark) and high salt concentration (350 mmol/L NaCl). These findings contrast with recent research on Arabidopsis, where AtPIF3 is identified as a negative regulator of salt tolerance through SOS2-mediated phosphorylation, which decreases its stability23. The differences in findings between maize and Arabidopsis can be attributed to several key factors. First, species-specific responses play a crucial role. The genetic and physiological differences between monocots (maize) and dicots (Arabidopsis) suggest that PIFs may have evolved distinct regulatory mechanisms in these plants. Second, the experimental conditions vary significantly. Our study's long day photoperiod and high salt concentration might induce different stress responses compared to those observed in other studies. Light signaling and stress pathways interact in ways that can significantly affect the stability and function of PIF proteins53. Additionally, regulatory mechanisms differ between species. The SOS2-mediated phosphorylation pathway in Arabidopsis might not function identically in maize. In Arabidopsis, this pathway destabilizes AtPIF3, reducing its negative impact on salt tolerance23. However, in maize, ZmPIF3 might interact with different proteins or pathways, leading to alternative regulatory outcomes. Furthermore, PIF proteins often exhibit functional redundancy22,53. In maize, other PIF family members might complement or counteract the role of ZmPIF3, influencing the overall stress response and leading to species-specific variations in PIF function and stress adaptation21,22.
Our study underscores the importance of considering species-specific regulatory networks and environmental conditions when studying the role of PIFs in abiotic stress responses. Future research should aim to dissect the precise molecular mechanisms underlying ZmPIF3 function in maize and compare them with those in Arabidopsis and other species. Additionally, exploring the interactions between PIFs and other stress-responsive pathways, such as hormonal signaling and ion homeostasis, could provide deeper insights into how plants balance growth and stress responses. Understanding these complex interactions is crucial for developing crop varieties with enhanced tolerance to salinity, a pressing need in the face of increasing soil salinization and global climate change. Our findings highlight the potential for targeted manipulation of PIF pathways to improve crop resilience and productivity under adverse environmental conditions.
In conclusion, this study provides compelling evidence of the critical role played by ZmPIF3.1 in enhancing salt tolerance in A. thaliana. Through detailed molecular characterization and functional analysis, we have demonstrated that overexpression of ZmPIF3.1 not only mitigates the effects of salt stress by enhancing proline accumulation and activating robust reactive oxygen species scavenging mechanisms but also modulates vital hormonal and calcium-signaling pathways. These regulatory modifications contribute significantly to the improved stress resilience observed in transgenic lines. Moreover, the transcriptional regulation activities of ZmPIF3.1, particularly its influence on transcription factors and the alternative splicing mechanism, underscore its potential as a key player in plant adaptive responses to environmental stresses. Looking forward, these findings highlight the importance of further exploring the molecular mechanisms and regulatory networks of ZmPIF3.1, which could pave the way for developing crop varieties with enhanced tolerance to salt and other stresses, thus contributing to sustainable agriculture and food security.
Data availability
The datasets generated during this study are available from the corresponding author upon reasonable request. All results generated during this study are included in this published article and its supplementary information files. The raw reads data is available at the NCBI Sequencing Read Archive (SRA; SUB14399439) database (Bioproject: PRJNA1104218).
References
Jiao, Y., Lau, O. S. & Deng, X. W. Light-regulated transcriptional networks in higher plants. Nat. Rev. Genet. 8, 217–230 (2007).
Franklin, K. A. & Whitelam, G. C. Light signals, phytochromes and cross-talk with other environmental cues. J. Exp. Bot. 55, 271–276 (2004).
Li, X., Liang, T. & Liu, H. How plants coordinate their development in response to light and temperature signals. Plant Cell 34, 955–966 (2022).
Kim, J. et al. Functional characterization of phytochrome interacting factor 3 in phytochrome-mediated light signal transduction. Plant Cell 15, 2399–2407 (2003).
Soy, J. et al. Phytochrome-imposed oscillations in PIF3 protein abundance regulate hypocotyl growth under diurnal light/dark conditions in Arabidopsis. Plant J. 71, 390–401 (2012).
Ni, M., Tepperman, J. M. & Quail, P. H. PIF3, a phytochrome-interacting factor necessary for normal photoinduced signal transduction, is a novel basic helix-loop-helix protein. Cell 95, 657–667 (1998).
Ni, M., Tepperman, J. M. & Quail, P. H. Binding of phytochrome B to its nuclear signalling partner PIF3 is reversibly induced by light. Nature 400, 781–784 (1999).
Laloum, T., Martín, G. & Duque, P. Alternative splicing control of abiotic stress responses. Trends Plant Sci. 23, 140–150 (2018).
Vogler, F., Schmalzl, C., Englhart, M., Bircheneder, M. & Sprunck, S. Brassinosteroids promote Arabidopsis pollen germination and growth. Plant Reprod. 27, 153–167 (2014).
Dantas, L. L. B. et al. Alternative splicing of circadian clock genes correlates with temperature in field-grown sugarcane. Front. Plant Sci. 10, 1614 (2019).
Kwon, Y. J., Park, M. J., Kim, S. G., Baldwin, I. T. & Park, C. M. Alternative splicing and nonsense-mediated decay of circadian clock genes under environmental stress conditions in Arabidopsis. BMC Plant Biol. 14, 136 (2014).
James, A. B. et al. Alternative splicing mediates responses of the Arabidopsis circadian clock to temperature changes. Plant Cell 24, 961–981 (2012).
Park, M. J., Seo, P. J. & Park, C. M. CCA1 alternative splicing as a way of linking the circadian clock to temperature response in Arabidopsis. Plant Signal. Behav. 7, 1194–1196 (2012).
Yadav, S., Irfan, M., Ahmad, A. & Hayat, S. Causes of salinity and plant manifestations to salt stress: A review. J. Environ. Biol. 32, 667–685 (2011).
Almeida, D. M., Oliveira, M. M. & Saibo, N. J. M. Regulation of Na+ and K+ homeostasis in plants: Towards improved salt stress tolerance in crop plants. Genet. Mol. Biol. 40, 326–345 (2017).
Geilfus, C. M., Ludwig-Müller, J., Bárdos, G. & Zörb, C. Early response to salt ions in maize (Zea mays L.). J. Plant Physiol. 220, 173–180 (2018).
Huqe, M. A. S. et al. Characterization of maize hybrids ( Zea mays L.) for detecting salt tolerance based on morpho-physiological characteristics, ion accumulation and genetic variability at early vegetative stage. Plants (Basel) 10, 2549 (2021).
Gao, Y. et al. A maize phytochrome-interacting factor 3 improves drought and salt stress tolerance in rice. Plant Mol. Biol. 87, 413–428 (2015).
Gao, Y. et al. A maize phytochrome-interacting factors protein ZmPIF1 enhances drought tolerance by inducing stomatal closure and improves grain yield in Oryza sativa. Plant Biotechnol. J. 16, 1375–1387 (2018).
Gao, Y. et al. Roles of a maize phytochrome-interacting factors protein ZmPIF3 in regulation of drought stress responses by controlling stomatal closure in transgenic rice without yield penalty. Plant Mol. Biol. 97, 311–323 (2018).
Gao, Y. et al. The phytochrome-interacting family of transcription factors in maize (Zea mays L.): Identification, evolution, and expression analysis. Acta Physiol. Plant 41, 8 (2019).
Kumar, I., Swaminathan, K., Hudson, K. & Hudson, M. E. Evolutionary divergence of phytochrome protein function in Zea mays PIF3 signaling. J. Exp. Bot. 67, 4231–4240 (2016).
Ma, L. et al. Phytochromes enhance SOS2-mediated PIF1 and PIF3 phosphorylation and degradation to promote Arabidopsis salt tolerance. Plant Cell 35, 2997–3020 (2023).
Zhang, X., Henriques, R., Lin, S. S., Niu, Q. W. & Chua, N. H. Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 1, 641–646 (2006).
Lv, D. W. et al. Proteomic and phosphoproteomic analysis reveals the response and defense mechanism in leaves of diploid wheat T. monococcum under salt stress and recovery. J. Proteom. 143, 93–105 (2016).
McGlincy, N. J. et al. Regulation of alternative splicing by the circadian clock and food related cues. Genome Biol. 13, R54 (2012).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Zou, P. et al. Low-molecular-weightt polysaccharides from Pyropia yezoensis enhance tolerance of wheat seedlings (Triticum aestivum L.) to salt stress. Front. Plant Sci. 9, 427 (2018).
Yamada, M. et al. Effects of free proline accumulation in petunias under drought stress. J. Exp. Bot. 56, 1975–1981 (2005).
Jin, M.-K. et al. ROS as a key player in quinolone antibiotic stress on Arabidopsis thaliana: From the perspective of photosystem function, oxidative stress and phyllosphere microbiome. Sci. Total Environ. 848, 157821 (2022).
Deinlein, U. et al. Plant salt-tolerance mechanisms. Trends Plant Sci. 19, 371–379 (2014).
Lozano-Juste, J., Alrefaei, A. F. & Rodriguez, P. L. Plant osmotic stress signaling: MAPKKKs meet SnRK2s. Trends Plant Sci. 25, 1179–1182 (2020).
Mittler, R., Zandalinas, S. I., Fichman, Y. & Van Breusegem, F. Reactive oxygen species signalling in plant stress responses. Nat. Rev. Mol. Cell Biol. 23, 663–679 (2022).
Schieber, M. & Chandel, N. S. ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 (2014).
Rodríguez, A. A. & Taleisnik, E. L. Determination of reactive oxygen species in salt-stressed plant tissues. Methods Mol. Biol. 913, 225–236 (2012).
Yu, Z. et al. How plant hormones mediate salt stress responses. Trends Plant Sci. 25, 1117–1130 (2020).
Zhao, S. et al. Regulation of plant responses to salt stress. Int. J. Mol. Sci. 22, 4609 (2021).
Ren, C. G., Kong, C. C. & Xie, Z. H. Role of abscisic acid in strigolactone-induced salt stress tolerance in arbuscular mycorrhizal Sesbania cannabina seedlings. BMC Plant Biol. 18, 74 (2018).
Huang, Y. et al. Salt stress promotes abscisic acid accumulation to affect cell proliferation and expansion of primary roots in rice. Int. J. Mol. Sci. 22, 10892 (2021).
Zheng, Y., Huang, Y., Xian, W., Wang, J. & Liao, H. Identification and expression analysis of the Glycine max CYP707A gene family in response to drought and salt stresses. Ann. Bot. 110, 743–756 (2012).
Quist, T. M. et al. HOS3, an ELO-like gene, inhibits effects of ABA and implicates a S-1-P/ceramide control system for abiotic stress responses in arabidopsis thaliana. Mol. Plant 2, 138–151 (2009).
Tian, Q., Uhlir, N. J. & Reed, J. W. Arabidopsis SHY2/IAA3 inhibits auxin-regulated gene expression. Plant Cell 14, 301–319 (2002).
Sano, T. et al. Calcium ions are involved in the delay of plant cell cycle progression by abiotic stresses. FEBS Lett. 580, 597–602 (2006).
Tang, R. J., Wang, C., Li, K. & Luan, S. The CBL-CIPK calcium signaling network: Unified paradigm from 20 years of discoveries. Trends Plant Sci. 25, 604–617 (2020).
Chen, L. et al. The Brassica napus calcineurin B-Like 1/CBL-interacting protein kinase 6 (CBL1/CIPK6) component is involved in the plant response to abiotic stress and ABA signalling. J. Exp. Bot. 63, 6211–6222 (2012).
Seo, P. J. et al. A self-regulatory circuit of CIRCADIAN CLOCK-ASSOCIATED1 underlies the circadian clock regulation of temperature responses in Arabidopsis. Plant Cell 24, 2427–2442 (2012).
Thines, B. & Harmon, F. G. Ambient temperature response establishes ELF3 as a required component of the core Arabidopsis circadian clock. Proc. Natl. Acad. Sci. U. S. A. 107, 3257–3262 (2010).
Fowler, S. G., Cook, D. & Thomashow, M. F. Low temperature induction of Arabidopsis CBF1, 2, and 3 is gated by the circadian clock. Plant Physiol. 137, 961–968 (2005).
Dong, M. A., Farré, E. M. & Thomashow, M. F. Circadian clock-associated 1 and late elongated hypocotyl regulate expression of the C-repeat binding factor (CBF) pathway in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 108, 7241–7246 (2011).
Jiang, B. et al. PIF3 is a negative regulator of the CBF pathway and freezing tolerance in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 114, E6695-e6702 (2017).
Li, Z. et al. Functions of phytochrome interacting factors (PIFs) in adapting plants to biotic and abiotic stresses. Int. J. Mol. Sci. 25, 2198 (2024).
Funding
This research was supported by grants from the National Natural Science Foundation of China (Nos. U1504315 and 32001565) and the Science and Technology Project in Henan Province (No. 212102110244).
Author information
Authors and Affiliations
Contributions
Z.H. designed the experiments and wrote the initial draft of the manuscript. L.T., H.L., X.Z., S.L., J.Z., S.G., B.W., L.Z., Y.J., and Y.G. actively prepared the materials, collected the data, and performed the analysis. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, Z., Liu, H., Zhao, X. et al. Functional characterization of maize phytochrome-interacting factor 3 (ZmPIF3) in enhancing salt tolerance in arabidopsis. Sci Rep 14, 19955 (2024). https://doi.org/10.1038/s41598-024-70427-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-70427-1
