
Abstract
Little is known about the mitochondrial genome of the family Eurybrachidae, with only two species sequenced. This study added one more mitogenome of Loxocephala sichuanensis in this family. The mitochondrial genome length of this species was 15,605 bp, consisting of 37 genes: 13 PCGs, 2 rRNAs, 22 tRNAs, and a control region. An unusually high A + T content, reaching 94.3% at the third codon position of 13 PCGs in Loxocephala, was found in Eurybrachidae, which was the highest among all planthoppers, especially on N-strand. Three tandem repeat regions were detected in the control region. Phylogenetic analyses based on complete mitochondrial genome sequences from 145 species (encompassing 18 planthopper families and 135 species in Fulgoromorpha as ingroup, and 6 other non-planthopper families in Auchenorrhyncha as outgroup) were conducted. Six datasets (PCG123R24, PCG123R2, PCG123, PCG12R24, PCG12R2, PCG12) were established to investigate the influence of 22 tRNAs and the third codon of the 13 PCGs of mitogenome for phylogeny analyses. Both Maximum likelihood and Bayesian trees supported the monophyly of the superfamilies Delphacoidea and Fulgoroidea. Delphacoidea, consisting of Cixiidae and Delphacidae as sister group, was in the basal position of Fulgoromorpha. In Fulgoroidea, the families Meenoplidae and Kinnaridae, Dictyopharidae and Fulgoridae, Acanaloniidae and Tropiduchidae were sister groups which were strongly supported. Caliscelidae was close to the sister group Lophopidae with Eurybrachidae. The four families Flatidae, Nogodinidae, Ricaniidae and Issidae were closely related. The position of Tettigometridae was uncertain. Derbidae and Achilidae form a sister group when 22 tRNAs were included in the phylogeny. The joining of the tRNA sequences of mitochondrial genome enhanced the stability of family-level nodes and adjusted some phylogenetic positions, highlighting the significant role of joining tRNAs in phylogenetic analyses. Including or excluding the third codon position of 13 PCGs generally did not affect the overall phylogenetic structures of Fulgoromorpha.
Similar content being viewed by others


Introduction
Fulgoromorpha, a captivating insect group within the order Auchenorrhyncha, consists of 21 extant families and over 14,000 species worldwide1. Previously classified all as Fulgoroidea Latreille, 1807 for recent taxa, it has recently been divided into two superfamilies: Delphacoidea Leach, 1815 and Fulgoroidea2,3. These attractive planthoppers are all phytophagous insects, adults sucking on leaves, stems and roots, while some nymphs sucking on the fluid in fungal hyphae4,5. They convert the unabsorbed sugars into a delicate substance called honeydew, which also inadvertently promotes the growth of mold, thus causing unexpected secondary damage to the plant6. Planthopper also serve as an important carrier of plant pathogens that spread plant diseases6,7. Consequently, several of them have become economic pests6,7.
The family Eurybrachidae Stål, 1862 is a relatively small group within the superfamily Fulgoroidea consisting of 40 genera and 210 species worldwide and divided into two subfamilies and six tribes1. The species Loxocephala sichuanensis Chou and Huang, 1985 is a particular and rare species found only in the western part of Sichuan, China in genus Loxocephala Schaum, 1850 of tribe Loxocephalini Schmidt, 1908 in subfamily Eurybrachinae Stål, 1862 of the family Eurybrachidae8. This species was initially described as a subspecies L. sinica sichuanensis Chou and Huang, 19859, however later re-evaluated and upgraded to species status based on male genitalia by Wang and Wang8. L. sichuanensis can be easily recognized by these morphological characteristics: tegmina tawny, basal 2/5 of the costal margin with a broad castaneous band, clavus area decorated with several white spots, apical part of tegmina interspersed with around 20 uniform black spots; the vertex, pronotum, and mesonotum are all tawny; frons is orange; legs are red8.
Previous studies in Eurybrachidae mostly focused on the description of new genera and species, but molecular studies are scarce, especially for mitochondrial gene studies. It is crucial to recognize that mitochondrial genes represent the most prevalent insect sequences in genetic data, and the entire mitochondrial genome plays a key role in various fields such as molecular systematics, population genetics, phylogeography, diagnostics, and molecular evolutionary studies10. So far, only 44 partial sequences obtained by Sanger sequencing from nine Eurybrachidae species are available on NCBI (https://www.ncbi.nlm.nih.gov/)11, while only two species Loxocephala perpunctata Jacobi, 1944 (GenBank accession number: MW848343) and Klapperibrachys cremeri (GenBank accession number: NC_084361) with complete mitochondrial gene sequenced and published in Eurybrachidae12,13.
In this study, the mitogenome of Loxocephala sichuanensis, a local endemic species in Sichuan, China, was added and analyzed in Eurybrachidae. A large dataset phylogeny in Fulgoromorpha, based on all the available mitochondrial genomes from NCBI, was conducted using both Maximum likelihood (ML) and Bayesian inference (BI) methods to explore the relationships within different families in planthoppers. Besides of the 13 protein coding (PCGs) genes and 2 ribosomal RNA (rRNA) genes of the mitogenome commonly used for phylogeny, the sequences of the 22 transfer RNA (tRNA) genes were also added to the phylogeny analyses which may improve the resolution and accuracy of the nodes10. Due to the high A or T bias in the third codon of the protein coding genes in the mitogenome, the inclusion or exclusion of the third codon of the 13 PCGs was also tested to examine its influence on the phylogeny of Fulgoromorpha. Finally, six different datasets (1. PCG123R24: 13 PCGs + 22 tRNAs + 2 rRNAs; 2. PCG123R2: 13 PCGs + 2 rRNAs; 3. PCG123: 13 PCGs; 4. PCG12R24: First and second codon positions of 13 PCGs + 22 tRNAs + 2 rRNAs; 5. PCG12R2: First and second codon positions of 13 PCGs + 2 rRNAs; 6. PCG12: First and second codon positions of 13 PCGs) were used for phylogenetic analyses in this study.
Results and discussion
Mitochondrial genome structure and composition of Loxocephala sichuanensis
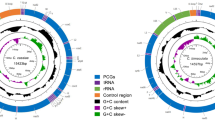
The complete mitochondrial genome length of L. sichuanensis was 15,605 base pairs (bp) (GenBank accession number: OR663919). It included 37 genes: 13 PCGs, 2 rRNAs, 22 tRNAs, and a control region (Supplementary file 1). This finding is comparable to other reported mitochondrial genomes in insects10. Most of the genes (9 PCGs, 14 tRNAs) were located on the J-strand, while the remaining genes (4 PCGs, 2 rRNAs, 8 tRNAs) were on the N-strand (Table 1). The mitogenome’s base composition was 46.5% adenine (A), 34.1% thymine (T), 12.1% cytosine (C), and 7.3% guanine (G), with an overall A + T content of 80.6%. This proportion is comparable to levels identified in other sequenced Eurybrachidae species12,13. Additionally, the genome displayed a positive AT skew (0.154) and a negative GC skew (− 0.245). The strong A + T bias may result from mutational pressure, a phenomenon also observed in other mitochondrial genomes in Hemiptera14.
The combined length of the 13 PCGs of L. sichuanensis was 10,945 bp. The overall PCGs exhibited a negative AT skew (− 0.119) and GC skew (− 0.021), while the PCGs on J-strand had a positive AT skew (0.056). The general A + T content in the third codon position of the 13 PCGs of the two Eurybrachidae species, L. sichuanensis and L. perpunctata, was significantly higher than in other Fulgoromorpha species (Fig. 1). Both species exhibited an A + T content of 94.3% in the third codon position, which was the highest among the 135 Fulgoromorpha species (Fig. 1). Additionally, the A + T content of the third codon on the N-strand was exceptionally higher than on the J-strand (Fig. 1). All PCGs started with codons ATN, except for nad5, which started with the codon GTG (Table 1). The four genes cox1, cox2, atp6 and cox3 used an incomplete T as the stop codon (Table 1), which may be converted to TAA through polyadenylation after transcription15.

The A + T content in the third codon position of 13 PCGs of 135 Fulgoromorpha species.
The total length of the 22 tRNAs of L. sichuanensis was 1,395 bp. The gene arrangement of tRNAs (Table 1) was the same as the other species L. perpunctata in Loxocephala12. Both species exhibited positive AT skew and GC skew for the total tRNA genes. The 22 tRNAs had only 18 bp differences between the above two species. Most of the tRNAs could be folded into the typical trilobal structure, except for tRNA-Ser1 and tRNA-Val, which were missing DHU stems (Supplementary file 2). The mismatched types in the secondary structure of tRNAs contained A-A, A-C, A-G and U-U, with A-A predominating.
The two rRNA genes, respectively located between trnL1 and trnV, and between trnV and the A + T rich region (Table 1), with a total length of 1,937 bp. They had a high A + T content (80.6%), a negative AT skew and a positive GC skew.
The control region, located between rrnS and trnI on the J-strand, was 1,192 bp (Table 1), with the A + T content reaching to 82%. It contained three tandem repeat regions: a 144 bp unit repeated 2.1 times, then a 16 bp unit repeated 2.1 times, and finally a 392 bp repeat region with the unit “TTTTTCCAAATTTTGTCACTT” repeated 18.7 times (Fig. 2) (Supplementary file 3). This occurrence of three repeats is uncommon in Fulgoromorpha, where most species have only one or two repeats16,17, but some species in Flatidae could reach to four18.

Organization of the A + T rich region of L. sichuanensis. The light yellow box, dark blue box and red box represent respectively the first, the second and the third repeat regions; the numbers in the box represent the number of sequence repetitions, and the uppercase letters indicate the repeated sequences.
L. sichuanensis had a total of 42 bp overlaps across 12 genes, with the longest overlaps (8 bp) occurring between trnW and trnC, and between nad6 and cytb (Table 1). The total length of intergenic spacers was 178 bp, with the longest spacer (163 bp) located between trnH and nad4 (Table 1).
Phylogenetic analyses of Fulgoromorpha
The tree topologies generated from six different data matrices (PCG123R24, PCG123R2, PCG123, PCG12R24, PCG12R2, PCG12) (Supplementary files 4–15) showed some variation among families in Fulgoromorpha but were generally consistent. Both Maximum likelihood (ML) and Bayesian inference (BI) analyses produced well-resolved phylogenetic trees with moderate to strong branch value support (Figs. 3, 4).

Phylogenetic tree produced from ML analysis. Numbers on branches are bootstrap (BS) support values, respectively for datasets PCG123R24, PCG123R2, PCG123, PCG12R24, PCG12R2, PCG12.

Phylogenetic tree produced from BI analysis. Numbers on branches are Bayesian posterior probabilities (PP) support values, respectively for datasets PCG123R24, PCG123R2, PCG123, PCG12R24, PCG12R2, PCG12.
In all the analyses under the site-homogeneous models, the ingroup Fulgoromorpha was always well supported as a monophyletic group with strong Bootstrap (BS) support in ML trees and strong posterior probability (PP) support in BI trees (Figs. 3, 4). The ingroup was separated into two large clades: the first clade (BS = 100, PP = 1) included the families Cixiidae and Delphacidae, representing the monophyletic superfamily Delphacoidea which recently separated from Fulgoroidea in 2022 and 2023 by Bourgoin and Szwedo2,3 according to the morphological characters; the second large clade (BS = 100, PP = 1) grouped the remaining 16 Fulgoromorpha families together, representing the monophyletic superfamily Fulgoroidea.
Within the superfamily Fulgoroidea, the Meenoplidae + Kinnaridae clade was consistently in the most basal position, with strong support (BS = 100, PP = 1), sister to the remaining Fulgoroidea families. The monophyly and sister relationship of Dictyopharidae and Fulgoridae were strongly supported (BS = 100, PP = 1) across all trees.
A noteworthy finding was the sister relationship between Derbidae and Achilidae, which was supported in all the BI trees and only in datasets that included 22 tRNAs in ML tree. These families were mostly sister to the Dictyopharidae + Fulgoridae clade, with strong support for their monophyly. The families Acanaloniidae and Tropiduchidae were consistently identified as sister taxa, forming a basal group (BS = 100, PP = 1) to all other families: Tettigometridae, Caliscelidae, Lophopidae, Eurybrachidae, Nogodinidae, Ricaniidae, Flatidae, and Issidae.
However, the position of Tettigometridae was uncertain within this clade. In Bayesian trees, Tettigometridae was positioned at the base of Lophopidae, Eurybrachidae, and Caliscelidae. In ML trees, this positioning was only observed in the PCG123R24, PCG123, PCG12R24, and PCG12 datasets. In the other two datasets (PCG123R2 and PCG12R2), Tettigometridae was sister to Flatidae.
Eurybrachidae, including L. sichuanensis, consistently formed a stable clade (Caliscelidae + (Lophopidae + Eurybrachidae)). This result verified the previous morphological study by Emeljanov19 and the molecular study based on 18S, 28S, H3 and Wingless genes for the sister group relationships of the families Eurybrachidae and Lophopidae20.
The last major clade in Fulgoroidea always comprised the four families Nogodinidae, Ricaniidae, Flatidae, and Issidae. The sister relationship between Nogodinidae and Ricaniidae received strong support in all analyses, except for the ML tree based on the PCG12R24 dataset, which showed the topology (Nogodinidae + (Ricaniidae + Flatidae)) with low support, indicating that it should not be considered. The observed topologies generally supported the arrangement (Flatidae + ((Nogodinidae + Ricaniidae) + Issidae)), unless rRNAs were excluded from the datasets. The monophyly of these four families were supported in this study.
The comparative phylogeny study for different matrixes
The analyses revealed that in the Bayesian tree topology which included the 22 tRNA sequences, the tree topology differed from the trees generated from other datasets. Notably, the inclusion of tRNA sequences resulted in a large clade with the sister group relationship of (Derbidae + Achilidae) and (Fulgoridae + Dictyopharidae), an unprecedented topology found for the first time in this study.
In younger branches, joining tRNA sequences significantly altered the topology, particularly within some branches of Issidae and Tropiduchidae. The addition of tRNA sequences also increased the stability of higher-level nodes. For example, the common node of Tettigometridae, Lophopidae, Eurybrachidae, and Caliscelidae showed higher PP values, 0.75 and 0.85 in datasets PCG123R24 and PCG12R24, respectively, compared to 0.66 and 0.76 in datasets without tRNA sequences (PCG123R2 and PCG12R2) (Figs. 3, 4). Similarly, the node summarizing showed that the (Nogodinidae + Ricaniidae + Issidae + Flatidae) cluster has PP values of 0.75 and 0.85 in the datasets PCG123R24 and PCG12R24, compared to 0.66 and 0.76 in the reduced tRNA datasets (PCG123R2, PCG12R2) (Figs. 3, 4). It is evident that a dataset comprising only 13 PCG sequences is insufficient to produce a topological structure similar to that of the trees obtained by incorporating tRNA or rRNA datasets21, such as the 19 nodes of the Issidae family in BI tree.
When comparing relationships across different datasets, we found that including two rRNAs and 22 tRNAs altered the position of the family Flatidae in Fulgoromorpha. It was no longer appeared as a sister group to Issidae when only 13 PCGs were used (datasets PCG123 and PCG12), but was positioned at the base of the ((Nogodinidae + Ricaniidae) + Issidae) clade in the other four datasets that included more genes, either contained tRNAs or rRNAs.
In the BI tree analyses, the structures of PCG12 and PCG123 are comparable, the same result occured when compared PCG12R2 to PCG123R2, and PCG123R24 to PCG12R24. This similarity indicates that incorporating the third codon position did not affect the overall structure. However, the inclusion of two rRNA genes altered the structural relationships in the PCG12 and PCG123 datasets, changing the topology from (Nogodinidae + Ricaniidae) + (Issidae + Flatidae) to Flatidae + ((Nogodinidae + Ricaniidae) + Issidae). The addition of 22 tRNA genes in the PCG123R24 and PCG12R24 datasets preserved this revised branch structure but modified the (Derbidae + Achilidae) + (Fulgoridae + Dictyopharidae) branch, which was not present in the earlier datasets.
In the ML tree analyses, Derbidae was positioned at the base of Achilidae or Fulgoridae + Dictyopharidae when only PCGs included. However, with the inclusion of 22 tRNA genes in the PCG123R24 and PCG12R24 datasets, its position shifted to a (Derbidae + Achilidae) + (Fulgoridae + Dictyopharidae) structure. The branch structure of Flatidae, Nogodinidae, Ricaniidae, and Issidae also changed when joining tRNAs. The structure (Nogodinidae + Ricaniidae) + (Issidae + Flatidae) in PCG123 and PCG12 datasets changed to Flatidae + ((Nogodinidae + Ricaniidae) + Issidae) in PCG123R24 and PCG12R24 datasets, and this new topology remained consistent regardless of the addition of rRNA or tRNA genes.
Conclusion
The mitogenome of Loxocephala sichuanensis in family Eurybrachidae from Fulgoroidea was sequenced, assembled and annotated. The structure of mitogenome for this species was analyzed. An unexpected finding was that the notably high A + T content at the third codon position of 13 PCGs, particularly on the N-strand of Loxocephala in Eurybrachidae within Fulgoromorpha. Phylogenetic analysis based on the mitogenomes from 145 species, including 18 planthopper families as ingroup and 6 outgroup families, was conducted within Auchenorrhyncha. The establishment of the superfamilies Delphacoidea and Fulgoroidea was confirmed. Relationships between the different families in Fulgoromorpha were explored. Adding 22 tRNAs into dataset showed a more stable result and higher node values in the phylogenetic analyses. Although some relationships within Fulgoromorpha are still uncertain, increasing the sample size and adding more molecular data may solve these issues in the future.
Materials and methods
Sample preparation, DNA extraction and sequencing
The specimen of L. sichuanensis was collected from the Wolong Nature Reserve in Sichuan Province, China. After collection, the specimen was promptly conserved in alcohol and maintained at a temperature of − 20 degrees at the China West Normal University, Sichuan, China. Following an accurate morphological identification under the stereomicroscope with photos of type specimen of L. sichuanensis in Wang and Wang8, the total genomic DNA was extracted from the thorax and legs of the specimen using an Animal Tissue DNA Extraction kit. The whole mitochondrial genome sequence was generated using high-throughput sequencing (Illumina NovaSeq6000 platform with insert size 400 bp and sequencing mode paired ends 2 × 150 bp).
Sequence assembly, annotation, and analysis
Raw paired reads were quality-trimmed (Q ≥ 20) and assembled using Geneious 2022.1.122. The mitochondrial genomes of L. perpunctata (MW848343)、Klapperibrachys cremeri (NC_084361)、Laodelphax striatellus (MK292916)、Zecheuna tonkinensis (MW872013) were chosen as reference sequences. The annotation of mitogenome was made by the same software with the same reference species. When annotating, the protein coding genes were identified by Geneious using Open Reading Frame, then checked if could translate to continuous proteins under the invertebrate genetic code; the tRNA genes were identified through the online websites tRNA Scan-SE server (http://lowelab.ucsc.edu/tRNAscan-SE/) and MITOS2 (http://mitos2.bioinf.uni-leipzig.de/index.py) and their secondary structure predicted meanwhile23,24; the rRNA genes and control region were identified by the boundaries of tRNA genes and the comparison with other mitogenomes in Fulgoromorpha. The mitogenome map was produced using Proksee (https://proksee.ca/)25. The A + T content of the third codon position for Fulgoromorpha (135 species) was made with software Origin. The mitogenomic information, nucleotide base composition, A + T/G + C content, AT skew/GC skew were analyzed with PhyloSuite26 (Supplementary file 16). Strand asymmetry was calculated in terms of formulae: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C)27. The secondary structure of tRNAs was manually mapped using Adobe Illustrator CS6 based on the structure predicted by MITOS2. Tandem repeats of A + T rich region was identified by Tandem Repeats Finder (https://tandem.bu.edu/trf/trf.basic.submit.html)28.
Phylogenetic analysis
The mitogenomes of 145 species from Auchenorrhyncha (Supplementary file 17) were used for phylogenetic analysis, of them, 135 Fulgoromorpha species (Delphacoidea and Fulgoroidea) as ingroup, while three species from Cercopoidea (representing by families Aphrophoridae and Cercopidae), two species from Cicadoidea (representing by family Cicadidae), and five species from Membracoidea (representing by families Membracidae, Aetalionidae & Cicadellidae) as the outgroup. Except for the species L. sichuanensis sequenced, assembled and annotated in this study, the rest mitogenomes were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/)11. The 13 PCGs, 22 tRNAs and 2 rRNAs from mitogenomes were extracted automatically using PhyloSuite26, then MAFFT29 aligned for each gene. The alignments were subsequently refined using the codon-aware program MACSE v2.0630 for 13 PCGs, ambiguously aligned fragments were removed in batches using Gblocks 0.91b31, gap sites were removed with trimAl v1.2rev5732, and then concatenated into different datasets. To investigate the influence of 22 tRNAs and the third codon of the 13 PCGs of mitogenome for phylogeny analyses, this study established six different data matrices:
-
1.
PCG123R24: 13 PCGs + 22 tRNAs + 2 rRNAs
-
2.
PCG123R2: 13 PCGs + 2 rRNAs
-
3.
PCG123: 13 PCGs
-
4.
PCG12R24: First and second codon positions of 13 PCGs + 22 tRNAs + 2 rRNAs
-
5.
PCG12R2: First and second codon positions of 13 PCGs + 2 rRNAs
-
6.
PCG12: First and second codon positions of 13 PCGs).
Modelfinder was used to identify appropriate partitions and select suitable models for each partition under the Bayesian information criterion (BIC)(Supplementary file 18).
Both Maximum likelihood (ML) and Bayesian inference (BI) methods were used for phylogenetic analyses based on the full mitochondrial genomes except for control region. Maximum likelihood analysis was performed by IQ-TREE33, using the Ultrafast bootstrap algorithm with 300,000 replicates, partitions with Edge-linked. Bayesian analysis was performed with MrBayes34. Two simultaneous runs were performed, each with one cold and three hot chains. Markov Chain Monte Carlo (MCMC) sampling estimated the posterior probability with 800,000 generations, sampling every 1000 generations once, with an average standard deviation of split frequencies below 0.05 and a relative burn-in rate of 25%, partitions with Edge-unlinked. Phylogenetic trees were visualized using the online website itol (https://itol.embl.de/)35.
Data availability
Data associated with this study has been deposited at NCBI under the accession number OR663919 (https://www.ncbi.nlm.nih.gov/nuccore/OR663919.1/). All data generated or analysed during this study are included in this published article and its Supplementary Information files.
References
Bourgoin, T. FLOW (Fulgoromorpha Lists on The Web): A World Knowledge Base Dedicated to Fulgoromorpha. Version 8. 2024. Available online: http://flow.hemiptera-databases.org/flow/ (Accessed on 4 Jan 2024).
Bourgoin, T. & Szwedo, J. Toward a new classification of planthoppers Hemiptera Fulgoromorpha: 1. What do Fulgoridiidae really cover?. Ann. Zool. 72, 951–962 (2022).
Bourgoin, T. & Szwedo, J. Toward a new classification of planthoppers Hemiptera Fulgoromorpha: 2. Higher taxa, their names and their composition. Zootaxa 5297, 562–568 (2023).
Huang, X.-D., Chen, X.-S., Yang, L. & Long, J.-K. Gigasanalis, a new genus of the tribe Achilini with the description of a new species from China (Hemiptera, Fulgoromorpha, Achilidae). Eur. J. Taxon. 852, 85–97 (2022).
Wilson, S. W. Keys to the families of Fulgoromorpha with emphasis on planthoppers of potential economic importance in the southeastern United States (Hemiptera: Auchenorrhyncha). Fla. Entomol. 88, 464–481 (2005).
Lee, D.-H., Park, Y.-L. & Leskey, T. C. A review of biology and management of Lycorma delicatula (Hemiptera: Fulgoridae), an emerging global invasive species. J. Asia-Pacific Entomol. 22, 589–596 (2019).
Urban, J. M. Perspective: Shedding light on spotted lanternfly impacts in the USA. Pest Manag. Sci. 76, 10–17 (2020).
Wang, M.-L. & Wang, Y.-L. Review of the genus Loxocephala Schaum, 1850 (Hemiptera: Fulgoromorpha: Eurybrachidae) with description of three new species from China. Zootaxa 3664, 176–198 (2013).
Chou, I., Wang, S.-Z. & Huang, J. Priskribo de novaj specioj de Fulgoroedoj el Cinio (Homopteroj: Fulgoroedoj). Entomotaxonomia 7, 29–38 (1985).
Cameron, S. L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Ann. Rev. Entomol. 59, 95–117 (2014).
Federhen, S. The NCBI taxonomy database. Nucl. Acids Res. 40, D136–D143 (2012).
Xu, S.-Y. & Chen, X.-S. Characterization of the complete mitochondrial genome of Loxocephala perpunctata (Hemiptera: Eurybrachidae). mtDNA, Part B. 7, 967–968 (2022).
Wang, W.-Q. et al. A phylogeny with divergence-time estimation of planthoppers (Hemiptera: Fulgoroidea) based on mitochondrial sequences. Zool. J. Linnean Soc. 201, 86–97 (2024).
Song, N., Liang, A.-P. & Ma, C. The complete mitochondrial genome sequence of the planthopper, Sivaloka damnosus. J. Insect Sci. 10, 76 (2010).
Ren, F.-G., Zhang, N., Zhang, L., Miller, E. & Pu, J. J. Alternative Polyadenylation: A new frontier in post transcriptional regulation. Biomarker Res. 8, 67 (2020).
Wang, W.-Q. et al. Characterization of the complete mitochondrial genomes of two species of the genus Aphaena Guérin-Méneville (Hemiptera: Fulgoridae) and its phylogenetic implications. Int. J. Biol. Macromol. 141, 29–40 (2019).
Zhang, H. et al. Comparative analysis of the complete mitochondrial genomes of five species of Ricaniidae (Hemiptera: Fulgoromorpha) and phylogenetic implications. Biology 11, 92 (2022).
Ai, D.-Q., Peng, L.-F., Qin, D.-Z. & Zhang, Y.-L. Characterization of three complete mitogenomes of Flatidae (Hemiptera: Fulgoroidea) and compositional heterogeneity analysis in the planthoppers’ mitochondrial phylogenomics. Int. J. Mol. Sci. 22, 5586 (2021).
Emeljanov, A. An attempt of construction of phylogenetic tree of the planthoppers (Homoptera, Cicadina). Entomol. Obozr 69, 353–356 (1990).
Urban, J. M. & Cryan, J. R. Evolution of the planthoppers (Insecta: Hemiptera: Fulgoroidea). Mol. Phylogenet. Evolut. 42, 556–572 (2007).
Bucher, M., Condamine, F. L., Luo, Y., Wang, M.-L. & Bourgoin, T. Phylogeny and diversification of planthoppers (Hemiptera: Fulgoromorpha) based on a comprehensive molecular dataset and large taxon sampling. Mol. Phylogenet. Evolut. 186, 107862 (2023).
Kearse, M. et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucl. Acids Res. 25, 955–964 (1997).
Donath, A. et al. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucl. Acids Res. 47, 10543–10552 (2019).
Grant, J. R. et al. Proksee: In-depth characterization and visualization of bacterial genomes. Nucl. Acids Res. 51, W484–W492 (2023).
Zhang, D. et al. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355 (2020).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evolut. 41, 353–358 (1995).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucl. Acids Res. 27, 573–580 (1999).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evolut. 30, 772–780 (2013).
Ranwez, V., Douzery, E. J., Cambon, C., Chantret, N. & Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evolut. 35, 2582–2584 (2018).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evolut. 32, 268–274 (2014).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucl. Acids Res. 49, W293–W296 (2021).
Acknowledgements
We sincerely thank the specimen collectors for their hard work in the field collection.
Funding
This study was supported by the National Natural Science Foundation of China (32100369).
Author information
Authors and Affiliations
Contributions
Shaobo FENG: Conceptualization, Data curation, Formal analysis, Visualization, Writing—original draft & Writing—review & editing. Menglin WANG: Conceptualization, Supervision & Writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Feng, S., Wang, M. Characterization of the complete mitochondrial genome of Loxocephala sichuanensis (Hemiptera: Eurybrachidae) with the phylogenetic analyses of Fulgoromorpha. Sci Rep 14, 21981 (2024). https://doi.org/10.1038/s41598-024-71948-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-71948-5

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}