
Abstract
Rice (Oryza sativa) is a staple food for billions of people across the globe, that feeds nearly three-quarters of the human population on Earth, particularly in Asian countries. Rice yield has been drastically reduced and severely affected by various biotic and abiotic stresses, especially pathogens. Controlling the attack of such pathogens is a matter of immediate concern as yield losses in rice crops could deprive millions of lives of nourishment worldwide. Pyricularia oryzae is one such pathogen that has been considered the major disease of rice because of its worldwide geographic distribution. P. oryzae belongs to the kingdom fungi, that causes rice blast ultimately adversely affecting the yield of the rice crop. Keeping in view this alarming scenario, the present study was designed so that the identifications of genome-encoded miRNAs of Oryza sativa were employed to target and silence the genome of P. oryzae. This study accomplished the computational analysis of algorithms related to miRNA target prediction. Four computational target prediction algorithms i.e., psRNATarget, RNA22, miRanda, and RNAhybrid were utilized in this investigation. The consensus among target prediction algorithms was created to discover six miRNAs from the O. sativa genome with the conservation of the target site fully evaluated on the genome of P. oryzae. The discovery of these novel six miRNAs in Oryza sativa paved a strong way toward the control of this disease in rice. It will open doors for further research in the field of gene silencing in rice. These miRNAs can be designed and employed in the future as experimentation to create constructs regarding the silencing of P. oryzae in rice crops. In the future, this research would be surely helpful for the development of P. oryzae resistant rice varieties.
Similar content being viewed by others


Introduction
Rice feeds over half the world's population, acting as a vital source of calories and energy, particularly in Asia and Africa1. Its reliable harvests and versatility ensure food security for millions, preventing malnutrition and promoting stability2. Rice is a valuable commodity, traded internationally and influencing global markets. Countries like Thailand, Vietnam, Pakistan, and India are major exporters, generating revenue and foreign exchange through the rice trade3. Rice isn't just for culinary delights, its byproducts, like bran and husk, find uses in animal feed, fuel, building materials, and even cosmetics4. Asia dominates the world's rice production and consumption, with a staggering 90% share, because of its warm and humid climate5,6,7,8. In Pakistan, it serves as a major source of food, income, and foreign exchange through export. Based on the data from the Economic Survey of Pakistan (2017–18), it is clear that the land devoted to growing rice has experienced a significant increase of 6.4%, rising from 2.74 million hectares in 2016–17 to an astonishing 2.89 million hectares. In addition, the production of rice has also seen a remarkable surge of 8.7%, resulting in an impressive yield of 7.44 million tons compared to 6.84 million tons. Rice output is expected to increase from 8.4 million tons to 9.3 million tons in 2021–22. The total production area of rice in Pakistan was 3,041,000 hectares, as per the Food and Agriculture Organization of the United Nations (FAO)6,9,10,11,12.
Pyricularia oryzae, also known as Magnoparthe oryzae, is a plant pathogenic fungus that poses a significant threat to rice crops worldwide13. The genomic information of Pyricularia oryzae varies among strains, and the fungus has been extensively studied through genome sequencing efforts. While the genome size can differ between isolates, researchers have identified key genomic features associated with pathogenicity, including regions housing virulence factors and effectors. According to data accessed from NCBI database (15 Jan. 2024) the genome of Pyricularia oryzae consists of 13,860 genes and 479,515 proteins. The genomic data has provided valuable insights into the molecular mechanisms underlying the fungus's ability to cause rice blast disease and interact with its host plant.
Pyricularia oryzae, is classified under the Sordariomycetes class in the Ascomycota phylum14. Pyricularia oryzae directly infects panicles and developing grains, causing them to become discolored, shriveled, and sterile, leading to substantial yield losses15. In severe cases, the entire panicle can be blighted, resulting in no grain formation at all. Pyricularia oryzae infects leaves, causing characteristic elliptical lesions that disrupt photosynthesis. This reduces the plant's ability to produce sugars for grain development, impacting yield. Severely infected leaves fall off prematurely, further limiting the plant's photosynthetic capacity and grain filling. Under favorable conditions for the fungus, rice blast can devastate entire fields, causing yield losses of up to 100%. Even in milder cases, yield reductions of 10–30% are common, posing a significant challenge for rice farmers and global food security. On the nutritional front, infected rice can show reduced protein content, a crucial nutrient for millions who rely on this staple. Blighted grains also tend to accumulate harmful mycotoxins, posing health risks.
This study aims to explore the potential of genome silencing approach in combating Rice blast and pave the way for a safer and more sustainable future. The utilization of genome silencing technology involves the application of RNAi to effectively cleave dsRNA into miRNAs that bind to mRNA, thereby impeding the translation of mRNA into protein16. The MicroRNAs also known as miRNAs, are small non-coding RNA molecules that play a crucial role in genome silencing. The miRNAs are approximately 21–25 nucleotides in length and are generated from hairpin-shaped RNA precursors. The miRNAs bind to complementary sequences of mRNA, which prevents mRNA from being translated into proteins17.
This study aims to predict Oryza sativa encoded miRNAs by employing in-silico approach. Four unique plant-based miRNA target prediction algorithms were utilized in this study. The miRNAs predicted by consensus of these four algorithms would be short-listed which can have the potential to silence the genome of Pyricularia oryzae. By the use of computational tools, we can predict the sites in the pathogenic fungal genome which would help control Rice blast. The predicted Oryza sativa miRNAs were further evaluated by estimation of free-energies, prediction of their secondary structure, site conservation analysis and also construction phylogenetic trees to study the evolutionary relationships among them.
Materials and methods
Extraction of miRNAs from miRBase
The first step was to collect data in the form of miRNAs from the public database. The miRNA data was collected from a primary data acquisition source which was miRbase https://www.mirbase.org/. Overall, 100 mature sequences of osa-miRNAs belonging to Oryza sativa were extracted from miRBase which were extracted and copied as separate files (http://www.mirbase.org/).
Retrieval of fungus genome from NCBI
The complete genome of Pyricularia oryzae with size 6358bps was retrieved from NCBI (Accession no. KY509032.1) and copied in another separate FASTA file (https://www.ncbi.nlm.nih.gov/).
Sequence analysis and ORF prediction of fungus
The genome sequencing of Pyricularia oryzae (P.oryzae) was done to explore various open reading frames (ORFs). The ORFs prediction of whole Pyricularia oryzae was executed by exploiting CLC Genomics Workbench (v23). The tools helped in correctly predicting ORFs and the order of nucleotides in Pyricularia oryzae genome highlighted in different colors to help in identification.
Target prediction
This research work was based on insilico approach of employing collections of Oryza sativa-encoded miRNAs to identify potential targets for silencing the genome of P. oryzae. A combination of four different target prediction algorithms, including RNA22, psRNATarget, RNA hybrid, and miRanda were exploited (Fig. 1). Predicting the targets for silencing the genome of Pyricularia oryzae involves identifying key miRNAs when silenced, could disrupt the fungus's growth and pathogenicity. Four miRNAs target prediction algorithms were utilized and Table 1 below shows the web sources and parameters opted for this prediction.

The Schematic diagram shows the flowchart describing the designed research work.
Target prediction by psRNATarget
psRNATarget is a computational tool for predicting the interactions between microRNA (miRNA), and its target messenger RNA (mRNA). This tool uses a sophisticated blend of techniques to effectively locate miRNA binding sites and their corresponding heteroduplexes18. Using the online web server, the Fasta sequences of Pyricularia oryzae and selected mature miRNAs were incorporated into the tool. Target sites of the rice miRNAs were identified with the user-defined settings with Expectation = 8, seed region 2-7nt, HPS size 19, gap open penalty = 2, and Gap extending penalty = 0.5 (https://www.zhaolab.org/psRNATarget/).
Target prediction by RNA22
RNA22 is a web server and this approach is used because of its unique characteristics to research new miRNA-mRNA interactions19. A pattern-based approach is used by RNA22 to recognize miRNA binding sites and the heteroduplexes that bind to them. When a miRNA attaches to the target RNA sequence, a complex called a heteroduplex is created. The maximum folding energy for RNA22 was set to 12 kcal/mol when it was run under default conditions (https://cm.jefferson.edu/rna22/Interactive/).
Target prediction by miRanda
The miRanda is a popular technique that integrates crucial components of miRNA-target prediction, including the conservation level and miRNA 3′UTR site. To locate miRNA binding sites in the 3′ untranslated region (UTR) of target mRNAs, miRanda employs a dynamic programming approach20. The parameters opted for analysis were Score threshold 14.00, MFE threshold of − 20.00 kcal/mol, gap open penalty of − 9.00, and gap extending penalty − 4.00 (http://www.microrna.org/microrna/getDownloads.do).
Target prediction by RNAhybrid
The RNAhybrid miRNA target prediction tool, used for both long and short miRNAs, the RNAhybrid algorithm was utilized to calculate the minimum free energy21. The Fasta sequences of Pyricularia oryzae genome and selected miRNAs sequences were incorporated in this tool for analysis, using mfe set at − 20.00 kcal/mol (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid).
Secondary structure prediction by RNAfold and RNA cofold algorithm
RNAfold web server is a publicly available server that predicts the secondary structure of single-stranded miRNA precursors with a high degree of accuracy, by uploading the heteroduplex sequences on the webserver. RNAcofold algorithm finds out the minimum free energy of the miRNA-mRNA duplexes. Using default settings each unique duplex pair was uploaded on the server for analysis (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAcofold.cgi).
Statistical and graphical analysis
In this study, graphical representations of tabulated data from miRNA target prediction were created by employing R programming. R offers a wide range of libraries and functions for creating various types of plots, including Venn diagrams. Readxl and ggplot2 were used in graph prediction22.
Mapping network interactions between miRNA and target fungus sequences
By utilizing the cutting-edge CIRCOS algorithm and Circos package v0.69-9R-Language, we were able to accurately map the interactions between predicted miRNAs and target fungus sequences with the help of a Circos plot23.
Site conservation analysis of miRNA- P. oryzae genome
Analyzing the site conservation through protein BLAST analysis, Pyricularia oryzae isolates' genome sequences were downloaded from NCBI and integrated into CLC Genomics Workbench. When aligning the sequences using the greatest precision alignment option, the gap open cost was set to 10.0 and the gap extension cost to 1.0. To look into the conservation of binding site locations, miRNA binding sites were located from the alignment.
Phylogenetic analysis of Pyricularia oryzae
To create a phylogenetic tree and analyze the evolutionary relationships between isolates, Pyricularia oryzae genomes underwent multiple sequence alignment (MSA) and were imported into the MEGA (Molecular Evolutionary Genetic Analysis) software24. The Neighbor-joining (with Bootstrap value 100) and maximum likelihood approaches were used to create a rooted phylogenetic tree from the aligned sequences.
Cloning of over-expressed miRNA gene construction
The specific gene sequences responsible for the production of mature miRNAs were synthesized and cloned in plant expression vectors. The miRNA synthesizing gene (bp) driven under polyubiquitin (Zm-Ubi) coupled with transgene enhancer regulatory elements (HYL1) was integrated into the plant expression vector, pCAMBIA1301 between kpnI and HindIII restriction sites by fermentas gene ligation kit (catalogue). The figure shows the gene construction map of the miRNA clone.
Transformation of highly activated synthetic miRNA gene in rice callus
The transformation of gene construction in rice was administered through callus formation. The callus in the rice variety was induced by employing sterilized rice seeds from the variety. The sterilization of rice seeds was executed through ethanol and bleach and rinsing was done thoroughly with autoclaved water. The rinsing was done time and again to ensure the complete removal of bleach from seed samples. The rice seeds were allowed to germinate on MS medium without hormones at dark conditions keeping a temperature of 25 °C for 3–4 days consecutively. The scutellum was cut from the germinated seeds and placed on callus induction medium (MS medium, 4.43 g/L supplemented with 2,4-D at 2–3 mg/L, and sucrose was added by 30 g/L solidified with agar) in sterile Petri dishes. The incubation of explants was implemented in the dark at 25 °C to induce callus formation which was typically observed within 2–3 weeks. Sub-culturing of the callus was also done every 2–3 weeks onto fresh MS callus medium to ensure optimum growth. The designed synthetic gene construct was obtained in highly pure DNA concentration through maxi-prep to obtain 5 μg/ml concentration. For further applications, transfer callus to regeneration or transformation media as needed, ensuring aseptic conditions throughout the process.
PCR confirmation of miRNA genes in putative transgenic rice lines
The extraction of DNA from rice plants was done through 2% CTAB method. Fresh leaves from control and putative transgenic plants were collected, frozen in liquid nitrogen, and ground into a fine powder. The 2% CTAB extraction buffer was added and incubated at 65 °C for 30 min. The chloroform:isoamyl alcohol with the correct ratio (24:1) was added and centrifuged to separate different phases. The aqueous phase was transferred to a new tube and cold isopropanol was added. Samples were incubated at − 20 °C for 30 min to allow precipitation of the rice DNA. The DNA pellet was obtained after centrifugation and the pellet was washed with 70% ethanol and air-dried. Finally, the DNA pellet was dissolved in TE buffer and the DNA quality was assessed by collecting concentrations. The putative rice transgenic plants with synthetic miRNA encoding gene construct were screened out with the help of primer specific PCR analysis. The primer sequences with forward, 3′-GCAGCACGTTTCTTTCTTGC-5′ and reverse primer GCGTTGGGAGCTTGTACTTC were utilized to amplify transgene in putative transgenic rice plants.
RT-PCR of control and transgenic rice lines
The RNA extraction from control and transgenic rice leaves was executed by TRIZOL method. The Real Time PCR of control and infected transgenic rice plants was done to show mRNA expression of Pyricularia oryzae and control plants. The primer sequence used in Real Time PCR was 5′-ACGAAACGGGATTATGCAAG-3′. The mRNA expression of control as well as transgenic plants was calculated as Ct values which were then processed through the Relative Expression Software Tool (REST) to originate quantifications of mRNA expression in different rice lines. The evaluations and expression analysis indicate mRNA expression of Pyricularia oryzae in various rice lines control as well as transgenic lines.
Ethics approval
Not applicable. All authors have read, understood, and have complied as applicable with the statement on “Ethical responsibilities of authors” as found in the Instructions for Authors and are aware that with minor exceptions, no changes can be made to authorship once the paper is submitted.
Ethical statement
It has been confirmed that the experimental data collection complied with relevant institutional, national, and international guidelines and legislation with appropriate permissions from authorities of the Department of Biotechnology, Knowledge Unit of Science, University of Management and Technology, Sialkot Campus, Punjab Pakistan.
Results
Sequence analysis and ORF of Pyricularia oryzae
The genome sequencing & ORF prediction of Pyricularia oryzae is depicted in Fig. 2 and Table 2, encompassing the sequences of nucleotides, and Open Reading Frames (ORFs). This information is crucial for comprehending the genetic makeup of this pathogenic fungus and its interactions with host plants (Fig. 3). Five ORFs were predicted in the genome of P. oryzae. ORFs are open reading frames within a DNA sequence, potentially code for proteins. Targeting the ORF can prevent the virus or pathogen from producing the proteins that it needs to survive and replicate (Fig. 4). Thus, it can lead to the death of the pathogen, or it can prevent the pathogen from causing disease.

The ORFs predicted in Pyricularia oryzae genome by. The genome (KY509032.1) has five ORFs distributed across it, each having different lengths, location and size.

Circular view of the Pyricularia oryzae genome (6358 bp) depicting its more active ORFs as shown in different arrows in various colours.

The Amino acid sequences of the five predicted ORFs on the genome of Pyricularia oryzae.
Target site predicted in four computational algorithms
Following graphs were generated by statistical data analysis using R (Fig. 5). Graphs show the predicted target sites in four different algorithms. The figures below represent the predicted target sites of Oryza sativa miRNAs in the Pyricularia oryzae genome by four algorithms (psRNATarget, RNA22, miRanda, and RNA hybrid) (Table 3). The colored dots show their respective target sites in the different regions in the genome of P. oryzae (Fig. 6).

Predicted target sites of Oryza sativa osa-miRNAs based on used four insilico miRNA targets. (A) Prediction of miRNA targets by psRNATarget (B) Prediction of miRNA targets by RNAhybrid (C) Prediction of miRNA targets by RNA22 (D) Prediction of miRNA targets by miRanda. The targets are represented by colored dots respectively.

The Circos plots showing rice encoded miRNAs targeting the Pyricularia oryzae ORFs. The above four Circos map shows the interaction between miRNAs targeting the five ORF for (a) RNA22, (b) psRNATarget, (c) miRanda, (d) RNAhybrid algorithms.
Graphical representation of consensus of predicted miRNAs
The tabular data generated by four distinct target prediction algorithms underwent a rigorous transformation process utilizing the RStudio package. The data was then skillfully translated into visually appealing graphical representations using the readxl and ggplot2 functions, with the inclusion of a Venn Diagram to further enhance the presentation of results (Fig. 7).

Venn diagram that displays the precise targeting of Pyricularia oryzae genome by miRNAs derived from Oryza sativa. The Venn diagram reveals the total number of Oryza sativa encoded miRNAs predicted by miRanda, psRNATarget, RNA22, and RNAhybrid. It shows that six miRNAs were common in all four algorithms, 35 were common in at least three algorithm & 52 miRNAs were common in two computational algorithms. The diagram emphasizes the rigorous selection criteria for miRNAs, where only those predicted by a consensus of four diverse algorithms were chosen. These miRNAs exhibit effective inactivation of the Pyricularia oryzae genome.
According to results of four target prediction algorithms, six miRNAs can be confidently predicted: osa-miR171a, osa-miR166d-5p, osa-miR166b-5p, osa-miR164b, osa-miR396c-3p, osa-miR169a (Fig. 8). These miRNAs were common in four algorithms, target binding site of each miRNA predicted in four tools is mentioned in Table 4. Apart from that 35 were such miRNAs that were common in at least three computational algorithms and 52 miRNAs were common in two algorithms.

Depiction of an Intersection plot, that gives a comprehensive overview of miRNA predicted, utilizing various bioinformatics algorithms. A color-coded key is provided alongside the plot for easy reference. Out of all the miRNAs predicted by four distinct computational algorithms, six miRNAs were identified as common to these computational algorithms. With the consensus of target prediction algorithms, the precise locations of the six targeted areas of Oryza sativa have been confidently predicted.
Predicted miRNAs have the potential to deactivate the genome of P. oryzae. The ORFs identified in P. oryzae are subject to inactivation by predicted miRNAs (Fig. 9). Specifically, ORF1 is targeted by osa-miR166b-5p, ORF2 by osa-miR164b and osa-miR166b-5p, ORF3 by osa-miR166b-5p, osa-miR169a, and osa-miR171a, ORF4 by osa-miR166d-5p and osa-miR169a, and ORF5 by osa-miR171a as depicted in Table 5. As a result, these ORFs are unable to translate into the resulting protein, ultimately leading to the silencing of the P. oryzae genome.

Graphs showing the six consensus miRNAs that were common in each tool (i.e. miRanda, RNA22, psRNATarget, and RNAhybrid) with their respective target-site.
Free energies (ΔG) associated with optimal miRNA-mRNA interaction
The predicted six miRNAs by consensus of four algorithms were further evaluated by free energies. Free-energy estimation was obtained by RNAcofold web server of Vienna Package and shown in the following Table 6.
Secondary structure of predicted miRNAs
The secondary structures of miRNA precursors were obtained with utmost accuracy and precision using the RNAfold web server. Predicting the secondary structure of miRNA plays a crucial role in genome silencing. Table 7 shows the folding structure of six miRNA precursors osa-miR164b, osa-miR166b-5p, osa-miR166d-5p, osa-miR169a, osa-miR177a, osa-miR396c-3p.
Conservation analysis of predicted target sites in P. oryzae genome
Our study revealed that conserved miRNA target sites are significantly concentrated at the 3′ UTR and 5′ end of protein-coding genes, particularly in those genes associated with protein synthesis pathways. Furthermore, we discovered that the conserved nucleotides are present among various P. oryzae strains. As a result, we were able to enhance the precision and reliability of predicted miRNAs for these strains based on this conservation trend (Fig. 10).


Analysis of miRNA- P. oryzae genome binding site conservation. Four colors (red, yellow, green, blue) indicate the four nitrogenous bases. Red indicates Adenine, Yellow indicates Guanine, and Green & Blue indicate Thymine & Cytosine. The red bars in the above alignment show the percentage of conservation of the binding site from 0 to 100. The degree of conservation of the binding site can then be assessed by calculating the percentage of residues that are identical or similar between the aligned sequences. Marked sequences in the above figures show 100% conservation. Regions of the binding site that are highly conserved are more likely to be important for binding to the predicted miRNAs and are therefore more likely to be functional and considered potential target sites for predicted miRNAs.
Phylogenetic analysis of Pyricularia oryzae strain
The phylogenetic tree of the genome of Pyriculria oryzae was constructed by using multiple sequence alignments of the isolated strain of the species (Figs. 11 and 12). To produce the most accurate phylogenetic trees, we utilized two robust methods: Maximum Likelihood and Neighbor-Joining (with Bootstrap value 100).

Phylogenetic analysis of P.oryzae taxa by Maximum Likelihood method. The branches of a phylogenetic tree represent the evolutionary lineages that lead to different organisms. The green dot in the figure shows the strain under study. The closer two organisms are on a phylogenetic tree, the more recently they shared a common ancestor.

Phylogenetic analysis of P. oryzae taxa by Neighbor-Joining method. The bootstrap value for Neighbor-joining was 100. The bootstrap values within a phylogenetic tree serve as an indicator of the frequency, out of 100, that the same branch is seen when regenerating the phylogenetic tree on a resampled data set. If we observe this occurrence 100 out of 100 times, it corroborates our findings. The branches of a phylogenetic tree represent the evolutionary lineages that lead to different organisms. The orange dot in the figure shows the strain under study. The closer two organisms are on a phylogenetic tree, the more recently they shared a common ancestor.
Results of trees show that, the closer the two species are on the tree, the more closely related they are. The length of the branches on the tree shows the amount of evolutionary time that has passed since the two species diverged. The nodes on the tree represent common ancestors. The nodes are points where two or more lineages diverge from each other. The more recent a node is, the more recent the common ancestor that it represents. The root is the point where all of the lineages on the tree converge.
Successful ligation and PCR confirmation
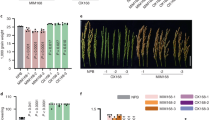
The successful ligation of transgene construct in plant expression vector was shown in 13A while PCR amilification of correponding gene was also represented in Fig. 13B

The schematic diagram of cloning strategy used for miRNA synthesizing genes in Rice (B). The PCR amplifications of Osdcl1 (gene) producing 561 bp gene product confirming gene transformation in rice.
Experimental validation of potential miRNAs through real time PCR analysis
The Real Time analysis was performed in both control and transgenic rice lines which were further shown in Fig. 14. The mRNA expression level in control rice plants was higher than over-expressed miRNA transgenic lines. The rice lines in control plants indicated that mRNA expression remained high as the corresponding miRNAs could not bind efficiently with fungal mRNA while hybridization of miRNA with mRNA in transgenic lines lead to degradation of corresponding transgenic miRNA lines. The obtained expressed data in Fig. 14 is the clear manifestation that those rice lines with enhanced biogenesis of mature miRNAs were successful in degradation of Pyricularia oryzae genome while control non transgenic lines showed increased level of fungus with high expression levels of mRNA. The trendline in the graph as indicated by dotted line represented the average mRNA expression of Pyricularia oryzae in enhanced miRNA rice lines.

The graphical demonstration of Pyricularia oryzae based mRNA expression in control and transgenic rice lines. The blue bars in the graph inidcated higher mRNA expression in lines while orange bars showed very lower mRNA expression in transgenic lines. (B) The (B) indicates and showed the difference between control and transgenic miRNA line which triggered gene silencing in rice against Pyricularia oryzae.
Discussion
Rice blast, a disease caused by a destructive fungus named Pyricularia oryzae, causes tremendous yearly loss to rice crops. The results of this study have successfully identified six miRNAs that have the potential to disrupt and inactivate the genome of P. oryzae. The discovery of these six miRNAs was made possible by utilizing four well-known computational algorithms. From the miRNAs predicted from these four tools, only those candidates were short-listed that were common in all four algorithms by using the intersection method. The six short-listed candidates were osa-miR171a, osa-miR166d-, osa-miR166b-5p, osa-miR164b, osa-miR396c-3p, osa-miR169a. These miRNAs were further evaluated by calculating their free energies, secondary structure prediction, and target-site conservation analysis. Moreover, phylogenetic trees were made to study the evolutionary relationship between the closely related isolates.
The results of this study have predicted six mature rice-encoded miRNAs that were expected to target the genome of P. oryzae towards developing the Rice blast disease in rice crops. The miRNAs identified in this study were predicted to interact with ORFs on the P. oryzae genome. Studies on miRNA target prediction for genome silencing have been extensive. Previous studies have reported the utilization of miRNA target prediction tools for silencing the genome of different pathogens25, examples include RYMV26, RTBV, RTSV27,28, CMD29, CLCuV30, MCMV31, ZYMV32. These studies involve computational approaches leveraging sequence complementarity, thermodynamic stability, and evolutionary conservation. No previous study was conducted on P. oryzae genome inactivation by using miRNA target prediction until now. However, there is a study published on the use of CRISPR/Cas 9 targeted knock-out of Rice susceptible genes. The results of this study supported that utilizing CRISPR/Cas9 to induce targeted mutations in rice susceptibility genes is a highly effective and innovative approach for developing resistance against Rice blast disease33.
In this study four insilico algorithms were utilized, i.e., psRNATarget18, RNA2219, miRanda20, and RNAhybrid21 with default parameters to establish an effective computational method for the validation of miRNA target prediction results. The findings indicate that the five ORFs predicted in the P. oryzae genome are highly susceptible to targeting by consensus miRNAs. Among the six miRNAs predicted by consensus of four algorithms, silencing of the P. oryzae genome is the direct result of the targeting of ORF1 by osa-miR166b-5p, ORF2 by osa-miR164b and osa-miR166b-5p, ORF3 by osa-miR166b-5p, osa-miR169a, and osa-miR171a, ORF4 by osa-miR166d-5p and osa-miR169a, and ORF5 by osa-miR171a. In other words, the inability of these ORFs to translate into resulting protein is a direct consequence of the aforementioned targeting, and there is no room for doubt in this regard.
Our study confirmed the previous findings emphasizing the crucial role of miRNAs in plant defense against pathogens. For example, research has demonstrated differential expression of miRNAs in rice in response to P. oryzae infection, with some of these miRNAs regulating defense-related genes34. Our study significantly advances previous findings by pinpointing specific miRNAs capable of targeting the P. oryzae genome, potentially boosting rice resistance. The study has identified six candidate miRNAs belonging to different miRNA families, each with unique functions in plant development and defense. For instance, the miR166 family is known to regulate class III homeodomain-leucine zipper genes, playing a significant role in plant development and stress responses34. In a similar vein, the miR164 family is known to target NAC transcription factors, which are crucial for plant defense against pathogens35. Based on our study, the presence of these miRNAs strongly indicates their influential role in regulating defense-related genes in rice, potentially leading to enhanced resistance against P. oryzae.
The free energy calculations and secondary structure analysis of these miRNAs conclusively demonstrate their potential to form stable complexes with their target sites. Calculating free energy is a crucial factor in determining the stability and efficacy of miRNA-mediated gene silencing. Our findings unequivocally demonstrate that all six miRNAs exhibit highly favorable free energy values, signifying robust interactions with their corresponding target sites in the P. oryzae genome. The consistently low free energy values strongly support the formation of stable complexes between these miRNAs and their target mRNAs, ultimately resulting in potent gene silencing. Furthermore, the target site conservation analysis strongly supports the idea that these miRNAs are conserved across different plant species, including rice, and have evolved to target specific genes involved in defense responses. In addition, the phylogenetic trees constructed using the Maximum Likelihood and NJ methods indicate that the closest relative is XM029898333.1, which is identified as the sister taxa of the strain KY509032.1.
Genome silencing by miRNA target prediction is a promising new approach to controlling rice blast disease. This approach can potentially be more effective and less harmful to the environment than other methods of controlling the disease36,37,38. The implementation of biological control using microbial agents with antimicrobial properties presents a remarkable alternative for managing Rice blast, however, it can be challenging to achieve in the field as its effectiveness is highly dependent on weather conditions39. The advantage of using genome silencing by miRNA target prediction to control rice blast disease is that it is a highly specific approach that can target specific genes involved in the pathogenesis of the disease40. Moreover, it is a relatively safe approach that does not involve the use of chemicals or other harmful substances41. Experimental validation remains crucial to confirm predicted targets due to the complexity of miRNA-target interactions and the need to minimize false positives.
Conclusion
This work confidently identified the potential candidate miRNAs that are most suitable for RNAi-mediated silencing of the P. oryzae genome. The identification is based on a consensus of plant miRNA target prediction methods. Further, experiments on wet laboratory provided strong evidence that these miRNAs were highly effective in controlling symptoms of Pyricularia oryzae in rice crop. Six miRNAs were predicted from the consensus of four algorithms: osa-miR171a, osa-miR166d-, osa-miR166b-5p, osa-miR164b, osa-miR396c-3p, osa-miR169a. These miRNAs were further evaluated by calculating their free energies, and secondary structures, and by thorough evaluation, it was revealed that the bond between miRNA-mRNA duplex is spontaneous and favorable. Site conservation analysis of P. oryzae isolates revealed the information about degree of conservation, and the higher percentage of conservation suggests the likelihood of potential target sites for predicted miRNAs. Moreover, phylogenetic analysis indicated the most closely related P. oryzae strains based on their evolutionary relationship.
Data availability
The complete genome of Pyricularia oryzae with size 6358bps was retrieved from NCBI (Accession no. KY509032.1) and copied in another separate FASTA file (https://www.ncbi.nlm.nih.gov/). The datasets used and/or analyzed during the current study available in the manuscript file.
Change history
08 November 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41598-024-77594-1
References
Zibaee, A. Rice: Importance and future. J. Rice Res. 1, e102 (2013).
Fukagawa, N. K. & Ziska, L. H. Rice: Importance for global nutrition. J. Nutr. Sci. Vitaminol. 65, S2–S3 (2019).
Muthayya, S., Sugimoto, J. D., Montgomery, S. & Maberly, G. F. An overview of global rice production, supply, trade, and consumption. Ann. N. Y. Acad. Sci. 1324, 7–14 (2014).
Ofongo, S. T., Kehraus, S., Iyayi, E. & Sudekum, K. in Tropentag 2008, Conference on International Research on Food Security. Natural Resource Management and Rural Development, Stuttgart-Hohenheim, Germany. 352.
Bandumula, N. Rice production in Asia: Key to global food security. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 88, 1323–1328 (2018).
Abbas, A., Rehman, A. & Javed, M. Exploring the potential of in vitro tissue culture in breeding programs of legume and pulse crops: Utilization and present condition. Bull. Biol. Allied Sci. Res. 2021, 36–36 (2021).
Rasheed, M. U., Malik, A. & Ali, M. S. Genetic variation and heritability estimates in chickpea seedling traits: Implications for breeding programs. Bull. Biol. Allied Sci. Res. 2024, 59. https://doi.org/10.54112/bbasr.v2024i1.59 (2024).
Rehman, K., Khalid, M. & Nawaz, M. Prevalence of potato leaf roll virus disease impacts and several management strategies to halt the damage. Bull. Biol. Allied Sci. Res. 2020, 21–21 (2020).
Chandio, A. A., Magsi, H. & Ozturk, I. Examining the effects of climate change on rice production: case study of Pakistan. Environ. Sci. Pollut. Res. 27, 7812–7822 (2020).
Junaid, M. D. & Gokce, A. F. Global agricultural losses and their causes. Bull. Biol. Allied Sci. Res. 2024, 66. https://doi.org/10.54112/bbasr.v2024i1.66 (2024).
Abbas, A., Rashad, A., Rehman, A. U. & Bukhari, M. S. Exploring the response mechanisms of rice to salinity stress. Bull. Biol. Allied Sci. Res. 2024, 58. https://doi.org/10.54112/bbasr.v2024i1.58 (2024).
Abbas, A., Arshad, A., Rehman, A. U., Bukhari, M. S. & Zaman, S. Revolutionizing plant breeding programs with advancements in molecular marker-assisted selection. Bull. Biol. Allied Sci. Res. 2024, 57. https://doi.org/10.54112/bbasr.v2024i1.57 (2024).
Talbot, N. J. On the trail of a cereal killer: Exploring the biology of Magnaporthe grisea. Annu. Rev. Microbiol. 57, 177–202 (2003).
Dubina, E. V. et al. Biodiversity of Pyricularia oryzae Cav. in rice-growing regions of the south of Russia using PCR method. Physiol. Mol. Biol. Plants 26, 289–303 (2020).
Simkhada, K. & Thapa, R. Rice blast, a major threat to the rice production and its various management techniques. Turk. J. Agric. Food Sci. Technol. 10, 147–157 (2022).
Coronnello, C. & Benos, P. V. ComiR: Combinatorial microRNA target prediction tool. Nucleic Acids Res. 41, W159–W164 (2013).
Riolo, G., Cantara, S., Marzocchi, C. & Ricci, C. miRNA targets: From prediction tools to experimental validation. Methods Protoc. 4, 1 (2020).
Dai, X., Zhuang, Z. & Zhao, P. X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 46, W49–W54. https://doi.org/10.1093/nar/gky316 (2018).
Loher, P. & Rigoutsos, I. Interactive exploration of RNA22 microRNA target predictions. Bioinformatics 28, 3322–3323 (2012).
Enright, A. et al. MicroRNA targets in Drosophila. Genome Biol. 4, 1–27 (2003).
Krüger, J. & Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34, W451–W454 (2006).
Gandrud, C. Reproducible Research with R and R Studio (Chapman and Hall/CRC, 2018).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547 (2018).
Gaafar, Y. Z. A. & Ziebell, H. Novel targets for engineering Physostegia chlorotic mottle and tomato brown rugose fruit virus-resistant tomatoes: In silico prediction of tomato microRNA targets. PeerJ 8, e10096 (2020).
Jabbar, B. et al. Target prediction of candidate miRNAs from Oryza sativa for silencing the RYMV genome. Comput. Biol. Chem. 83, 107127 (2019).
Tripathy, R. & Mishra, D. A computational approach of rice (Oryza sativa) plant miRNA target prediction against tungro virus. Procedia Eng. 38, 1357–1361 (2012).
Mohamed, N. A. et al. Candidate miRNAs from Oryza sativa for silencing the rice tungro viruses. Agriculture 13, 651 (2023).
Ashraf, M. A., Ali, B., Brown, J. K., Shahid, I. & Yu, N. In silico identification of cassava genome-encoded microRNAs with predicted potential for targeting the ICMV-Kerala begomoviral pathogen of cassava. Viruses 15, 486 (2023).
Ashraf, M. A., Brown, J. K., Iqbal, M. S. & Yu, N. Genome-Wide identification of cotton microRNAs predicted for targeting cotton leaf curl kokhran virus-Lucknow. Microbiol. Res. 15, 1–19 (2023).
Iqbal, M. S. et al. In silico MCMV silencing concludes potential host-derived miRNAs in maize. Front. Plant Sci. 8, 372 (2017).
Shahid, M. N., Rashid, S., Iqbal, M. S., Jamal, A. & Khalid, S. In silico prediction of potential mirnas to target zymv in cucumis melo. Pak. J. Bot. 54, 1319–1325 (2022).
Távora, F. T. et al. CRISPR/Cas9-targeted knockout of rice susceptibility genes OsDjA2 and OsERF104 reveals alternative sources of resistance to Pyricularia oryzae. Rice Sci. 29, 535–544 (2022).
Li, Y. et al. Multiple rice microRNAs Are involved in immunity against the blast fungus Magnaporthe oryzae. Plant Physiol. 164, 1077–1092. https://doi.org/10.1104/pp.113.230052 (2013).
Guo, W. et al. High-throughput sequencing and degradome analysis reveal neutral evolution of Cercis gigantea microRNAs and their targets. Planta 243, 83–95. https://doi.org/10.1007/s00425-015-2389-y (2016).
Min, H. & Yoon, S. Got target? Computational methods for microRNA target prediction and their extension. Exp. Mol. Med. 42, 233–244 (2010).
Fatima, S. et al. The genome-wide bioinformatics analysis of 1-aminocyclopropane-1-carboxylate synthase (acs), 1-aminocyclopropane-1-carboxylate oxidase (aco) and ethylene overproducer 1 (eto1) gene family of fragaria vesca (woodland strawberry). Bull. Biol. Allied Sci. Res. 2023, 38–38 (2023).
Li, Y. et al. The roles of rice microRNAs in rice–Magnaporthe oryzae interaction. Phytopathol. Res. 1, 1–12 (2019).
Sella, L. et al. Sustainable methods to control Pyricularia oryzae, the causal agent of rice blast disease. In Innovations in Land, Water and Energy for Vietnam’s Sustainable Development, 67–82 (2021).
Ekimler, S. & Sahin, K. Computational methods for microRNA target prediction. Genes 5, 671–683 (2014).
Roberts, J. T. & Borchert, G. M. Computational prediction of microRNA target genes, target prediction databases, and web resources. In Bioinformatics in microRNA Research, 109–122 (2017).
Acknowledgements
The authors are thankful to the Deanship of Scientific Research, King Khalid University, Abha, Saudi Arabia for Financially supporting this work through a large research Group Project under grant no. R.G.P. 2/369/44.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
T.S. carried out research work and wrote the initial draft of the manuscript. M.F.A. and S.A. conducted the data analysis. M.F.S., Q.A., and S.I. planned and supervised the study and edited the final version of the manuscript. M.Y.A., M.A.J. and Q.A. technically reviewed and finalized the draft. All authors reviewed the final version of the manuscript and approved it for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: In the original version of this Article author Mohammad Y. Alshahrani was incorrectly affiliated with “Department of Plant Breeding and Genetics, Faculty of Agriculture Sciences, University of the Punjab, P.O BOX. 54590, Lahore, Pakistan.” and the acknowledgement section had an error in the grant number mentioned. Full information regarding the correction can be found in the correction notice published with this article.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Suddal, T., Awan, M.F., Ali, S. et al. Target prediction of potential candidate miRNAs from Oryza sativa to silence the Pyricularia oryzae genome in rice blast. Sci Rep 14, 21813 (2024). https://doi.org/10.1038/s41598-024-72608-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-72608-4

{kind=link}