
Abstract
Cortisol is established as a reliable biomarker for stress prompting intensified research in developing wearable sensors to detect it via eccrine sweat. Since cortisol is present in sweat in trace quantities, typically 8–140 ng/mL, developing such biosensors necessitates the design of bioreceptors with appropriate sensitivity and selectivity. In this work, we present a systematic biomimetic methodology and a semi-automated high-throughput screening tool which enables rapid selection of bioreceptors as compared to ab initio design of peptides via computational peptidology. Candidate proteins from databases are selected via molecular docking and ranked according to their binding affinities by conducting automated AutoDock Vina scoring simulations. These candidate proteins are then validated via full atomistic steered molecular dynamics computations including umbrella sampling to estimate the potential of mean force using GROMACS version 2022.6. These explicit molecular dynamic calculations are carried out in an eccrine sweat environment taking into consideration the protein dynamics and solvent effects. Subsequently, we present a candidate baseline peptide bioreceptor selected as a contiguous sequence of amino acids from the selected protein binding pocket favourably interacting with the target ligand (i.e., cortisol) from the active binding site of the proteins and maintaining its tertiary structure. A unique cysteine residue introduced at the N-terminus allows orientation-specific surface immobilization of the peptide onto the gold electrodes and to ensure exposure of the binding site. Comparative binding affinity simulations of this peptide with the target ligand along with commonly interfering species e.g., progesterone, testosterone and glucose are also presented to demonstrate the validity of this proposed peptide as a candidate baseline bioreceptor for future cortisol biosensor development.
Similar content being viewed by others

Introduction
Non-invasive and unobtrusive detection of various health biomarkers such as cortisol is of active research interest1,2,3,4. These biosensors intended for cortisol detection via sweat5,6,7,8,9,10 offer a clear advantage over other biofluids such as saliva and urine for both non-invasive and periodic monitoring. Human eccrine sweat is a biomarker-rich fluid that offers some correlations with blood serum11,12. The possibility of detecting such analytes via non-invasive methods using readily available sweat can be made possible by designing these bioreceptors for specific analytes. The advent of nano-biosensing13,14,15,16 combined with various electrochemistry methods and artificial intelligence to interpret the results has made it possible to realize such biosensors. Ongoing research on such point-of-care sweat-based sensors is targeted towards improving their sensitivity and selectivity by testing various bioreceptor candidates.
The most common problems affecting the performance of such biosensors are cross-reactivity of the biorecognition element, stability of the capture molecules, ability to function in presence of interference from other species, etc. To overcome these challenges, researchers continuously strive to optimize the design of the biological recognition element i.e., bioreceptors. In fact, the design of effective biosensors primarily relies on identifying the most appropriate bioreceptor for the target ligand. Design of the bioreceptor needs to consider multiple parameters such as binding affinity towards the target ligand, specificity, selectivity, ease of synthesis, cost of synthesis, stability, sequence length and tertiary structure. Traditionally, some of these considerations are addressed by simply employing well-validated antibodies as the bioreceptor during biosensor design17,18,19. However, antibodies are large macromolecules that are generally expressed via living organisms. They also have other limitations such as temperature sensitivity, requirement of cold storage and lack of reusability. Hence, there is a strong requirement to come up with alternative bioreceptors.
In contrast, peptides are emerging as a promising class of bioreceptors as they are much smaller compared to antibodies and can be synthesized easily without a living organism. For example, the baseline peptide proposed in this manuscript is ten times shorter in length compared to the corresponding natural corticosteroid binding globulin (CBG) proteins 2V95 (rat) and 2VDY (human)20. We present a systematic in silico methodology for developing this peptide. We start by funnelling all the prospective candidate proteins for cortisol from the protein data bank21 to a semi-automatic high-throughput screening tool based on molecular docking (using AutoDock Vina version 1_1_2) for computing their interactions with cortisol. Subsequently, we rank these proteins according to their scores and perform full atomistic steered molecular dynamics (SMD) simulations using the classical molecular dynamics (MD) tool GROMACS22. The top three candidate proteins from Auto Dock Vina docking simulations are then selected to validate their free energy and dynamics by including the solvent effects present in an eccrine sweat environment25,26,27. The potential of mean force (PMF) calculation serves as a measure of the binding energy of these proteins with cortisol. The MD simulations are performed using GROMACS while the PMF is calculated via umbrella sampling and weighted histogram analysis method (WHAM)23.
Subsequently, a contiguous sequence of amino acids from the active binding site24 of the top candidate protein is selected as the potential peptide bioreceptor based on its structure and interactions. This selected peptide is then modelled to compare its docking performance with the parent protein, as well as its binding with interfering molecules such as progesterone, testosterone, and glucose. Finally, the peptide is modified with a cysteine residue at its N-terminus25,26 to facilitate attachment to a gold surface for transduction and future development of an electrochemical biosensor. Thus, our workflow aids in silico development of biosensors by initially screening a larger number of candidate protein receptors by docking, followed by selecting the right peptide sequence from the candidate proteins, and finally validating the peptide bioreceptor using MD simulations. This method can help computationally address the general challenges involving sensitivity and selectivity during bioreceptor development and identify one or two candidates that can be later experimentally verified.
Theory and method
Rationale for in silico bioreceptor design
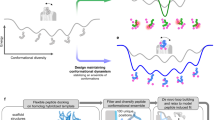
We follow a systematic in silico methodology for integration of multiple computational techniques such as semi-automated molecular docking using AutoDock Vina27,28 and MD simulations using GROMACS to design a baseline candidate peptide bioreceptor for cortisol. This combination approach enables us to enhance the search space of the candidate protein and conduct rapid high-throughput screening by studying their ligand binding interactions using docking, before validating the peptide using MD simulations. The workflow followed for the baseline peptide design is shown in Fig. 1.

Workflow of development of baseline peptide bioreceptor for cortisol.
The proposed in silico approach for modelling the baseline peptide bioreceptor can be divided into two steps: (1) molecular docking and (2) MD modelling. Molecular docking is performed for rapid screening of a large number of candidate proteins. Docking tools do not provide any information about the thermodynamics of the process or explicitly include appropriate solvent effects. Since MD is computationally intensive, it is subsequently used for validation of a limited number of candidates screened by molecular docking. We then employ a classical MD simulation technique known as ‘umbrella sampling’ to calculate the free energy profile of a specific ligand along the reaction coordinates for understanding the energy landscape of the process. The first step in umbrella sampling is to define a reaction coordinate that characterizes the process of interest. We choose the distance between the respective centre-of-mass of the protein and the ligand to track interactions and binding events. This is achieved by conducting an SMD pull simulation in the desired direction by implementing an imaginary spring. This is termed as the ‘constant velocity pull simulation’ since this rate is fixed once we fix the force constant of the spring. Subsequently, we calculate the potential of mean force (PMF) along this chosen reaction coordinate. The PMF represents the free energy as a function of the reaction coordinate. Multiple umbrella sampling simulations are performed with the system restrained along the reaction coordinate with each window. A harmonic biasing potential is applied to keep the system at a specific value of the reaction coordinate. The SMD simulations are executed in each umbrella window, allowing the system to explore its configurational space while remaining constrained along the reaction coordinate. The trajectories are collected for each window. Subsequently, a histogram of the reaction coordinate values is constructed based on the collected data. This histogram provides information about the probability distribution of the system along the reaction coordinate for reconstructing the PMF. Finally, using statistical mechanics principles, the PMF is reconstructed from the histogram data. This involves applying the weighted histogram analysis method (WHAM) to obtain the free energy profile as a function of the chosen reaction coordinate. The resulting PMF can be used to understand the energetics of the process. It reveals energy barriers, transition states, and stable states along the reaction pathway. Hence, umbrella sampling is employed to quantify the energy landscape and estimate the free energy changes for estimating the PMF.
Details of molecular docking
AutoDock Vina is the molecular docking tool employed in this work. Fifty candidate protein files are first selected from the protein data bank21 (PDB). These proteins are selected via keywords for binding to “cortisol”. The selection is guided by literature data for native corticosteroid-binding globulins (CBG) from various organisms such as rats, pigs, and humans. Proteins with > 75% similarity scores evaluated via BLAST scores are set aside as duplicates. Finally, thirty-three candidates are chosen for feeding to AutoDock Vina.
These PDB files are pre-processed to remove the water molecules from the complexes and modified by adding missing hydrogen atoms and Kollman charges. The processed files are stored as PDBQT files. The cortisol (ligand) PDB file is also similarly processed. The protein is placed in a grid box of approximately (120 Å)3 to enclose the entire protein. The ligand is then placed in its vicinity about 5 Å away from the protein and blind docking is performed. The binding configuration and binding energy of the protein-cortisol complex are obtained by minimizing the potential energy of the system. The level of exhaustiveness selected is 8. Figure S1 in supporting information shows the complete docking with the candidate protein 2V95. We rank the candidate proteins based on their binding affinity using the scoring function. Figure S2 in supporting information has the candidate proteins, their PDB IDs, binding energies and interaction diagrams. The purpose of this step is to identify the key interactions of the target analyte with the candidate bioreceptors. These candidate proteins then serve as a basis for identifying a shorter peptide sequence.
Details of MD simulations
System preparation
The top three protein and ligand complexes are selected from the list of screened proteins based on their interaction energies with cortisol, as listed in Table 1. Figure 2 shows the PDB ID, rendered structure and binding energy of the top three candidate proteins. Each model consists of the protein molecule complexed with the cortisol ligand, solvated by water molecules and 50 mM NaCl. The protein .pdb files, selected from the PDB database, are individually placed in a simulation box of approximately (10 nm)3 cube along with a single cortisol molecule in an eccrine sweat model30 represented by approximately 50 mM of NaCl. Additionally, sodium ions are added to ensure the system is charge neutral. The system is created as a protein-ligand complex and solvated in eccrine sweat solution model using CHARMM-GUI31. The force field used here has been CHARMM36 force field32,33,34,35 at a physiological temperature of 310 K. All simulations were performed in GROMACS 2022.6.
Simulation protocol
Each system is simulated via the following protocol. First, the energy of a single macromolecule is minimized in vacuum using the steepest descent algorithm (step size = 0.01 nm). This energy-minimized molecule is then solvated with water. The water molecules are modelled using a three-point water model (TIP3P)36. The biomolecules are systematically equilibrated by performing energy minimization, NVT and NPT simulations. The NVT and NPT simulations are done in two stages. In the first stage, position restraints are placed on the biomolecules and water is allowed to move freely. In the second stage, the simulation is performed without any restraints.
1 ns of simulation is performed at each stage except for the last one, where unrestrained NPT simulation is performed for 10 ns (the equilibration run). The simulation temperature and pressure are set at 310 K and 1 bar, respectively. After the equilibrium run, the system undergoes 100 ns of NPT simulation as a production run. The extracted trajectories are used to calculate properties. During the equilibration run, the temperature and pressure are controlled with the Berendsen37 thermostat and barostat with appropriate time constants.
In the production run, the temperature and pressure are controlled by the Nosé–Hoover thermostat38,39 and the Parrinello–Rahman barostat40, applying time constants of 2 ps and 5 ps, respectively. The electrostatic and van der Waals cutoffs are set at 1.2 nm. The particle mesh Ewald (PME) summation method is used to treat long-range electrostatic interactions. The neighbour list cutoff was set at 1.2 nm and updated at every tenth step. The simulations are performed for 10 ns and trajectories are saved every 100 ps. The protein is position restrained and the pull simulation conducted by aligning it along with positive Z axis which was selected as the reaction coordinate.
Umbrella sampling and PMF
The binding energy computation via umbrella sampling and potential of mean force (PMF) is employed to validate the binding affinity of the selected protein. The PMF is an indirect measure of the binding affinity of the biomolecules. To calculate the PMF, initial configurations are generated by constraining the ligand molecule at various locations from the protein using a harmonic potential. Starting configurations are generated by pulling the ligand molecule radially outwards from the protein, until the ligand (i.e. cortisol) is approximately 4 nm away from the protein. It is pulled at a constant velocity of 0.01 nm/ps. During this process, snapshots of the system are captured when the distance between the centre-of-mass of the protein and the ligand changes by 0.2 nm. The twenty captured windows are equilibrated for 1 ns, followed by 5 ns of NPT simulation using an umbrella harmonic constraint of 1000 kJ mol− 1 nm− 2. The simulation trajectory is saved every 10 ps. The PMF is generated using the weighted histogram analysis method23 (WHAM), as implemented using the gmx wham command in GROMACS. The binding energy (ΔG) is the difference between the highest and the lowest values of the PMF curve, given the values of the PMF converging to a stable value at a large distance i.e., selected reaction coordinates, as shown in Fig. 4. All these simulations are performed on AMD EPYC 7532 with 64 cores per node and 12 compute nodes at 2.4 GHz clock speed for approximately 15 days per SMD simulation to finally compute the PMF.
Identifying the baseline peptide
The active binding sites of the top candidate proteins are analysed and contiguous sequences of amino acids amongst these candidates are iteratively assessed to arrive at a baseline peptide while retaining its existing scaffold. The binding energies and the interaction diagrams of all thirty-three candidate proteins are illustrated in Figure S2 in the supporting information. The candidate protein 2V95 is assessed for selecting the most appropriate continuous sequence of amino acids. Different sequences of different length are selected, and among those, only one is chosen based on its length and its binding affinity. The candidate peptides are identified by selecting various continuous lengths from the active binding sites of protein 2V95 to arrive at the proposed baseline peptide as presented in Table S2. Only the protein 2V95 is selected over the other candidate proteins (listed in Table 1) due to its relatively shorter (< 50 amino acids long) sequence length of the binding site (ranging from residues 220 to 257) as compared to the other proteins. Each sequence length, ranging from 36 to 46 amino acids, is assessed for its binding affinity and selected based on a pre-determined binding affinity value for the shortest sequence length.
Table S1 lists different physicochemical properties of the selected peptides computed using PepCalc41. A comparison of the final selected peptide and other candidates in terms of sequence length and binding energies is presented in Table S2. Candidate #7 (proposed baseline peptide) offers a good binding score for the selected length of amino acids. Next, interference with three competing biomolecules (e.g. progesterone, testosterone and glucose) are assessed. Finally, the N-terminal is selected to incorporate a cysteine residue to promote attachment to a gold surface, keeping in mind future development as a biosensor. This cysteine residue is selected from an already existing residue in the native protein sequence of 2V95.
Validation of baseline peptide
The selected baseline peptide is then similarly modelled using GROMACS to assess its root mean square deviation (RMSD) and binding energetics via umbrella sampling. Its PMF is computed and compared with native protein which serves as a benchmark. The stability of the peptide is assessed via the RMSD values and binding energy via PMF and compared with native protein 2V95.
Results
High throughput screening via molecular docking and scoring
We screen a number of the candidate proteins from the protein database which can bind with cortisol and present them along with their binding energies via an AutoDock Vina based tool. The binding affinity results of top five candidate PDBs using molecular docking is listed in Table 1.
The interaction diagrams of the top three candidate proteins are presented in Fig. 2. These candidates were selected for further MD calculations and validation by performing umbrella sampling and computing the potential of mean force in presence of a solvent. 6NWL and 2VDY were not considered despite its higher binding energy as its binding pocket was hidden and > 100 amino acids long.

AutoDock Vina screening of top three candidate proteins with their corresponding interaction diagrams with cortisol.
The remaining list of thirty candidate proteins, considered along with their binding affinity, is presented for completeness in Figure S2. These candidate proteins were subsequently ranked according to their binding affinity. A continuous sequence of amino acids was selected as candidate bioreceptors based on their active binding pockets. This biomimetic procedure enables rapid selection of bioreceptors as compared to ab initio design of peptides via computational peptidology42.
Validation of the top candidates via steered molecular modelling
We performed SMD simulation of the top protein–ligand complex in eccrine sweat solution43. The results are presented for the evolution of the potential energy and RMSD of the native protein 2V95 and cortisol complex. Figure 3 shows the RMSD values, while Fig. 4 shows the binding affinity. The RMSD values after stabilization are within 0.6 nm which demonstrates the stability of the protein 2V95. We treat the native protein and cortisol complex as a benchmark in our work.
Finally, we employed WHAM, for extracting the PMF. The values of the PMF converge to a stable value at approximately 4 nm on the selected reaction coordinate as shown in Fig. 4. The binding energy (ΔG) is then simply the difference between the highest and the lowest values of the PMF curve. The difference between the highest and lowest values of PMF is approximately -10.2 kcal/mol. The RMSD of the native protein 2V95 is presented in supporting information Figure S5A and other candidate proteins are similarly modelled and presented in supporting information figures S5B and S5C. The results presented in supporting information in figures S10A and S10B for represent similar binding energy and serve as a validation for the selected SMD methodology and umbrella sampling settings.

RMSD of 2V95 native protein bound with cortisol over 100ns demonstrating stability.

PMF of 2V95 native protein with cortisol demonstrating binding force over pull distance i.e. reaction coordinate.
A close inspection of the energetically favourable binding conformations of the native protein (2V95) bound to cortisol offers a physically contiguous sequence of amino acid residues (from 220 to 257) as opposed to the other candidate proteins and therefore selected for further consideration for peptide design.
Protocol for selection of the baseline peptide and comparison with corticosteroid-binding globulins (CBGs)
The candidate bioreceptor peptide is identified by selecting a few contiguous sequences of amino acids (Table S2 in supporting information) from the active binding sites of the 2V95 protein, with the imposed constraint that each sequence should be less than 50 amino acids in length. The pre-determined binding affinity value with cortisol was imposed as another constraint as depicted in Figure S6 in supporting information. The selected sequences are listed with their corresponding binding energy with cortisol and compared with CBGs. Finally, a peptide with a sequence of 38 amino acids is selected with a cysteine residue at its end. It is represented by the single letter sequence CQLIQMDYVGNGTAFFILPDQGQMDTVIAALSRDTIDR. The selection ensures a good binding with cortisol with a relatively smaller sequence thereby, ensuring ease of synthesis at a lower cost.
Baseline peptide binding energies with competing species
The baseline peptide is then modelled with glucose, progesterone and testosterone. The binding energies of these interfering species are listed in Table 2. Figure 5 shows the interaction diagram of progesterone and glucose with cortisol. It was important to explore the binding affinity of the selected peptide with progesterone since CBGs are known to bind well with this hormone20. As we see, the binding energies of the peptide with all three of these species are much lower than the binding energy with cortisol.

Interaction diagram of interfering species such as progesterone and glucose with cortisol.
Similarity analysis of the baseline peptide using smart BLAST
The baseline peptide is further compared with the proteins 2V95 (371 amino acids long) and 2VDY (373 amino acids long), which are rat and human CBGs respectively. These two proteins were chosen for comparison due to their relatively higher binding affinity with cortisol and the presence of a physically contiguous sequence of interacting amino acids in the binding region. The comparison was performed using the Basic Local Alignment Search Tool (BLAST)44,45 tool hosted by the National Center for Biotechnology Information (NCBI) online server. The native peptide was found to have > 75% similarity with these two proteins, especially in their active binding sites for cortisol. The relevant screenshots from smart BLAST demonstrating similarity between baseline peptide and protein 2V95 and other candidate proteins are shown in Figure S3A and Figure S3B respectively in supporting information. When the selected baseline peptide is subsequently compared with other proteins from the database smart BLAST, it is observed that there is “landmark match” with both CBGs from as illustrated in Figure S3B. This is an extremely promising result considering our biomimetic route of peptide selection as opposed to ab initio peptide design via various extremely computationally intensive combinatorial methods25. The structures of these two large proteins have evolved to cater to a large number of design requirements, resulting in macromolecular structures that are more than 370 amino acid sequences long. Furthermore, the natural design considerations for a CBG and that of a bioreceptor are different. We argue that the entire macromolecular structure of the native proteins is not necessary for biosensor applications. In this work, we propose a lean peptide design approximately one tenth of the sequence length of native CBG proteins with comparable binding affinity and tertiary structure as a baseline candidate to develop the intended biosensor. The design considerations for our proposed biosensor are limited to bioreceptor development. Therefore, we have only focussed on parameters such as binding affinity with target ligand, solubility, sequence length, tertiary structure, and ability to bind with gold electrodes.
Significance of the in silico eccrine sweat model
The choice of the solvent model, namely the eccrine sweat model30 developed earlier by the same team, has a significant impact on determining the stability and strength of the protein-ligand complexes. The hydrophobic regions41 of the baseline peptide minimizes its exposure to water molecules by binding to ligands which can lead to increased binding affinity. Similarly, the hydrogen bonding capabilities influence the formation and strength of hydrogen bonds between proteins and ligands. Figure S4 shows the hydrophobic and hydrophilic parts of the peptide. Finally, as discussed in the next section, a full atomistic molecular dynamics simulation of the peptide is performed in eccrine sweat solution as a validation exercise.
Validation of the baseline peptide via steered molecular dynamics (SMD) simulation
We performed SMD simulation of the baseline peptide–ligand complex in an eccrine sweat environment as described in the methods section. Figure 6A shows the peptide anchored to gold in a sweat environment. The C-alpha atoms RMSD (< 0.6 nm) of the baseline peptide and ligand confirms the stability of the complex and the equilibration of the system as shown in Fig. 6B. Despite the considerable shorter sequence length, the baseline peptide is stable and retains bound cortisol for the entire simulation run of 30 ns. The binding energy (approximately – 9.8 kcal/mol) computed is comparable with the native protein 2V95 (approximately – 10.2 kcal/mol) and inspires confidence in the utility of the proposed baseline peptide.

(A) Model of the proposed baseline peptide bioreceptor immobilized on gold substrate and bound with cortisol docked in the binding cavity. (B) Binding pocket of baseline peptide and bound cortisol.

PMF plot with standard deviation of candidate baseline peptide bioreceptor with cortisol demonstrating binding affinity over pull distance of approx. 4 nm.

C-alpha atoms RMSD of candidate baseline peptide bound with cortisol demonstrating stability over 100 ns.
As seen in Fig. 7, the PMFs of the native protein 2V95 bound to cortisol and of the baseline peptide demonstrates binding affinity of approximately − 9.8 kcal/mol and in Figure S9 in supporting information demonstrates number of hydrogen bonds providing insights into the binding affinity over a 100 ns simulation.
Advantages of peptide bioreceptors over conventional antibody based bioreceptors
Traditional methods of developing biosensors involve using antibodies as the bioreceptor. Antibody based electrochemical sensors have been used for the measurement of cortisol and other biomolecules due to their high specificity and sensitivity. However, they suffer from limitations such as storage requirement, temperature instability, high cost, cross-reactivity, and batch-to-batch variability. Antibodies are large molecules that are not readily synthesized and can be chemically unstable17,19. To address the issues, we propose the development of an inexpensive, synthetic peptide which can be considered as an alternative to these antibodies. Our approach is biomimetically inspired i.e., selecting a continuous sequence of amino acids from the native protein, aimed at retaining structure, scaffold and stability of the finalized baseline peptide as opposed to combinatorial peptide design46.
Discussion
Our goal was to systematically arrive at a candidate baseline peptide following a biomimetic approach. In addition to considering the specific binding affinity towards cortisol and a shorter sequence length, other factors such as ease of immobilization on gold electrodes, solubility, ease of synthesis and cost of synthesis need to be considered. Furthermore, the CBGs presented in Table S2 demonstrate a strong binding affinity towards progesterone, which needs to be corrected in the proposed peptide design to avoid non-specific interactions.
Additional parameters such as size and sequence length of the peptide also play an important role in the design. In the current work, we have computed these values and have only optimized the design for the minimum continuous sequence length for the desired binding affinity. This is illustrated in the Table S2 (with peptides #1, #2, and #3 as the representative candidates) where various lengths of amino acid sequences are assessed before finalizing the 38 amino acids long sequence. The other native proteins that have similar or improved binding affinity with cortisol (e.g. 6NWL as presented in Table 1 and illustrated in Fig. 2) do not offer a contiguous sequence of amino acids < 50 AA, to create a peptide with a similar structure. Hence, they are not considered further. A final consideration is the ability of the designed peptide to bind to the surface of gold electrodes. This is achieved by a cysteine termination at the N terminal of the baseline peptide. The cysteine residue contains thiol groups that can readily form strong covalent bonds with gold atoms on the electrode surface, creating a stable gold-thiol bond.
The proposed peptide sequence CQLIQMDYVGNGTAFFILPDQGQMDTVIAALSRDTIDR, as depicted in Figure S4 in supporting information, is > 50% hydrophobic in nature. As it is an acidic peptide, it will require an acidic solvent to dissolve it initially. Later the solution pH will need to be adjusted to 6.3 to mimic the mean pH value of eccrine sweat. Preliminary simulations of the proposed peptide, as presented in Figure S7 and Figure S8 in supporting information, have shown that the immobilization of the peptide on gold does not significantly alter its structure. The RMSD of the peptide and cortisol is within reasonable limits as presented in Fig. 8. Finally, the RMSD plot of the peptide in Fig. 8 demonstrates no significant increase in RMSD as compared to the RMSD of the native protein 2V95, despite the shorter sequence of the peptide. The proposed baseline peptide is one tenth the size of the native protein. Hence estimated cost of synthesis for 5 mg of the peptide at > 80% purity (HPLC Purification) is approximately USD 400. In contrast, the same amount of a conventional monoclonal antibody for cortisol (e.g. CORT-1) costs USD 4000. Furthermore, the central part of the sequence demonstrates hydrophobicity to improve its affinity and stability. The cysteine amino acid at N-Terminal with a reactive - SH group provides ease of binding with gold electrodes. In summary, the proposed baseline peptide can be considered as an efficient, cost effective, and a viable alternative to antibody-based cortisol bioreceptors.
Summary
This work describes a systematic biomimetic approach to arrive at a baseline peptide bioreceptor for cortisol with the future goal of developing a biosensor. The baseline peptide bioreceptor is achieved by screening candidate proteins from the protein database via semi-automated AutoDock Vina based molecular docking. Subsequent explicit MD calculations serve as a validation to compute the PMF of the candidate shortlisted proteins. Finally, identifying the active binding sites made up of continuous sequences of amino acids amongst the top candidates are iteratively assessed to arrive at a baseline peptide sequence. Parameters such as binding energy, sequence length, tertiary structure, and cost of synthesis are considered. Physicochemical properties such as solubility, pH, and isoelectric point are also computed to arrive at the presented peptide. This peptide was then modelled to establish comparable binding energy values with the native proteins. We also explore its interference with competing biomolecules such as progesterone, testosterone and glucose. Additionally, the N-terminal is selected as cysteine residue (present in the native protein 2V95) which serves as a thiol bond with the gold electrode substrates for further development as a biosensor. The in silico eccrine sweat model earlier reported by us serves as a solvent to simulate the performance of a large number of candidate receptors for sweat biosensors and reduce the need for multiple laboratory experiments.
This work is intended to serve as a tool to aid the design of candidate bioreceptors for systematic development of various wearable biosensors. This high-throughput screening methodology can also serve as a horizontal means for design of the bioreceptor element of wearable sweat sensors for biomarkers other than cortisol. This tool can be used to compare various candidate peptides via their binding energies, simulate different sequence lengths, predict tertiary structures and interference with competing species. Thus, this design tool can serve as an aid to expedite the development of de novo biosensors.
Ethics statement
The entire data presented in this publication is simulated data and no human/animal tests were conducted.
Data availability
The data for simulations used from the protein data bank and high-throughput screening tool developed that support this study can be made available upon reasonable request from the corresponding author.
References
Bandodkar, A. J., Ghaffari, R. & Rogers, J. A. Don’t sweat it: The Quest for wearable stress sensors. Matter. 2, 795–797 (2020).
Singh, A., Kaushik, A., Kumar, R., Nair, M. & Bhansali, S. Electrochemical Sensing of Cortisol: a recent update. https://doi.org/10.1007/s12010-014-0894-2
Rice, P. et al. CortiWatch: watch-based cortisol tracker. Future Sci. OA5, (2019).
Cheng, C. et al. Battery-free, wireless, and flexible electrochemical patch for in situ analysis of sweat cortisol via near field communication. Biosens. Bioelectron.https://doi.org/10.1016/j.bios.2020.112782 (2021).
Russell, E., Koren, G., Rieder, M. & Van Uum, S. H. M. The detection of cortisol in human sweat. Ther. Drug Monit.https://doi.org/10.1097/ftd.0b013e31829daa0a (2013).
Bariya, M., Nyein, H. Y. Y. & Javey, A. Wearable sweat sensors. Nat. Electron.1, 160–171 (2018).
Hauke, A. et al. Complete validation of a continuous and blood-correlated sweat biosensing device with integrated sweat stimulation. Lab. Chip. 18, 3750–3759 (2018).
Ganguly, A., Rice, P., Lin, K. C., Muthukumar, S. & Prasad, S. A combinatorial Electrochemical Biosensor for sweat Biomarker Benchmarking. SLAS Technol.25, 25–32 (2020).
Kinnamon, D., Ghanta, R., Lin, K. C., Muthukumar, S. & Prasad, S. Portable biosensor for monitoring cortisol in low-volume perspired human sweat. Sci. Rep.7, (2017).
Torrente-Rodríguez, R. M. et al. Investigation of Cortisol Dynamics in human sweat using a graphene-based Wireless mHealth System. Matter. 2, 921–937 (2020).
Nyein, H. Y. Y. et al. Regional and correlative sweat analysis using high-throughput microfluidic sensing patches toward decoding sweat. Sci. Adv.5, (2019).
Yin, H. et al. ENGINEERING Regional and correlative sweat analysis using high-throughput Microfluidic sensing patches toward decoding sweat. Sci. Adv.5 (2019). http://advances.sciencemag.org/
Dutta, G. Nanobiosensor-based diagnostic system: transducers and surface materials. in Nanobiomaterial Engineering: Concepts and Their Applications in Biomedicine and Diagnostics 1–13 (Springer Singapore, doi:https://doi.org/10.1007/978-981-32-9840-8_1. (2020).
Rapini, R. et al. NanoMIP-based approach for the suppression of interference signals in electrochemical sensors. Analyst. https://doi.org/10.1039/c9an01244c (2019).
Murase, N., Taniguchi, S. I., Takano, E., Kitayama, Y. & Takeuchi, T. A molecularly imprinted nanocavity-based fluorescence polarization assay platform for cortisol sensing. J. Mater. Chem. B. 4, 1770–1777 (2016).
Munje, R. D., Muthukumar, S., Panneer Selvam, A. & Prasad, S. Flexible nanoporous tunable electrical double layer biosensors for sweat diagnostics. Sci. Rep.https://doi.org/10.1038/srep14586 (2015).
Demuru, S. et al. Antibody-coated wearable Organic Electrochemical transistors for Cortisol Detection in human sweat. ACS Sens.7, 2721–2731 (2022).
Zhou, Y., Hu, W., Peng, B. & Liu, Y. Biomarker binding on an antibody-functionalized biosensor surface: the influence of surface properties, electric field, and coating density. J. Phys. Chem. C. 118, 14586–14594 (2014).
Arya, S. K., Chornokur, G., Venugopal, M. & Bhansali, S. Antibody functionalized interdigitated µ-electrode (IDµE) based impedimetric cortisol biosensor. Analyst. 135, 1941–1946 (2010).
Klieber, M. A., Underhill, C., Hammond, G. L. & Muller, Y. A. Corticosteroid-binding globulin, a Structural Basis for Steroid Transport and proteinase-triggered release. J. Biol. Chem.282, 29594–29603 (2007).
RCSB PDB. Homepage. https://www.rcsb.org/
Abraham, M. J. et al. High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2. GROMACS, 19–25 (2015).
Rosenberg, J. M. et al. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. (1992). https://www.researchgate.net/publication/200147620
Badhe, Y., Gupta, R. & Rai, B. In silico design of peptides with binding to the receptor binding domain (RBD) of the SARS-CoV-2 and their utility in bio-sensor development for SARS-CoV-2 detection. RSC Adv.11, 3816–3826 (2021).
Johnson, S. et al. Surface-immobilized peptide aptamers as probe molecules for protein detection. Anal. Chem.80, 978–983 (2008).
Estrela, P. et al. Label-free sub-picomolar protein detection with field-effect transistors. Anal. Chem.82, 3531–3536 (2010).
Trott, O., Olson, A. J., AutoDock & Vina Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.31, 455–461 (2010).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python Bindings. J. Chem. Inf. Model.61, 3891–3898 (2021).
Lee, J. et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 Additive Force Field. J. Chem. Theory Comput.12, 405–413 (2016).
Deshpande, P., Ravikumar, B., Tallur, S., Paul, D. & Rai, B. Development of an insilico model of eccrine sweat using molecular modelling techniques. Sci. Rep.12, 20263 (2022).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem.29, 1859–1865 (2008).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature Methods 2016 14:1 14, 71–73 (2016).
Vanommeslaeghe, K. et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all‐atom additive biological force fields. J. Comput. Chem.31, 671–690 (2010).
Vanommeslaeghe, K. et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem.31, 671–690 (2009).
Huang, J. & MacKerell, A. D. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem.34, (2013).
Mark, P. & Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A105, (2001).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys.81, 3684–3690 (1984).
Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys.52, 255–268 (1984).
Nosé, S. & Klein, M. L. Constant pressure molecular dynamics for molecular systems. Mol. Phys.50, 1055–1076 (1983).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys.52, 7182–7190 (1981).
Lear, S. & Cobb, S. L. Pep-Calc.com: a set of web utilities for the calculation of peptide and peptoid properties and automatic mass spectral peak assignment. J. Comput. Aided Mol. Des.30, 271–277 (2016).
Zhou, P. & Huang, J. Computational Peptidology. Methods in Molecular Biology vol. 1268 http://www.springer.com/series/7651
Deshpande, P., Ravikumar, B., Tallur, S., Paul, D. & Rai, B. Eccrine Sweat Molecular Model for Development of de novo Biosensors. in 44th Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC) 914–917 (IEEE, 2022). doi: (2022). https://doi.org/10.1109/EMBC48229.2022.9871988
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res.50, D20–D26 (2022).
Boratyn, G. M. et al. Domain Enhanced Lookup Time Accelerated BLAST (2012).
Woolfson, D. N. A Brief History of De Novo Protein Design: Minimal, Rational, and Computational: De novo protein design. Journal of Molecular Biology vol. 433 Preprint at (2021). https://doi.org/10.1016/j.jmb.2021.167160
Acknowledgements
P.D, D.D, Y.B and B.R acknowledge support by Tata Consultancy Services (Research). High performance computing was carried out using the HPC cluster of Tata Consultancy Services. The authors acknowledge Prof. Ajay Panwar from IIT Bombay and Dr Rajgopal Srinivasan and Mr. Dharmendr Kumar from TCS Research for useful technical discussions.
Author information
Authors and Affiliations
Contributions
P.D has developed and written the entire manuscript as part of his PhD work with molecular docking simulations support from D.D and molecular modelling simulation support from Y.B The entire work is carried out under the guidance from PhD guides Prof S.T, Prof D.P and Industry guide Dr B.R All authors have reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deshpande, P., De, D., Badhe, Y. et al. An in silico design method of a peptide bioreceptor for cortisol using molecular modelling techniques. Sci Rep 14, 22325 (2024). https://doi.org/10.1038/s41598-024-73044-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73044-0
