
Abstract
The combination of Josiphos-type ligands with Pd catalysts has been instrumental in the rapid development of efficient catalytic processes. We performed density functional theory (DFT) calculations to elucidate the mechanisms and dynamic conformational changes responsible for the reactivity and selectivity observed in Pd-catalysed bicyclization/carbonylation of 1,6-enynes. DFT calculations indicated that the most favourable reaction pathway involves an unusual alkene insertion into the carbon–palladium bond to give high level of enantioselectivity. Here, the reactivity is enhanced by the self-adaptation of the Josiphos-Pd backbone, which allows for two distinct ligand conformations with different steric environments. A half-chair conformation is preferred in migratory insertion, which is both the rate-determining step and the enantioselectivity controlling step. The less hindered steric environment of the half-chair conformation allows for rapid migratory insertion, as confirmed by Surface distance projection maps and IGM analysis. Furthermore, IGM analysis shows that the steric effect between the phenyl group in the ligand and the methyl group on the allene of the substrate is important for enantioselectivity control.
Similar content being viewed by others

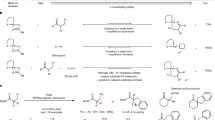

Introduction
Chiral biphosphines are important ligands with a privileged backbone and they have been widely used in many metal-catalysed asymmetric reactions1,2,3,4,5,6,7,8,9. Generally, a wide range of organic synthesis transformations can be achieved through the electronic properties and steric effects induced by chiral biphosphine ligands bound to metal centres, as well as chiral biphosphine–substrate noncovalent interactions10,11,12,13. Josiphos-type14,15,16 chiral ligands are representative of this type of ligand. They feature ferrocene backbones and often exhibit both planar chirality and asymmetric C-stereocenters, making them attractive options for asymmetric catalysis. Josiphos ligands can combine with a metal to form a six-membered cyclic backbone, which can have two different conformations (Fig. 1a), i.e., the boat conformer and the half-chair conformer, which have both been confirmed by X-ray crystal structure analysis17,18. In the asymmetric reaction, to improve reactivity and selectivity, does the conformation of the six-membered Josiphos-metal skeleton change? With this question in mind, we explored the conformational changes19,20,21 between the different conformations of A and B during the catalytic cycle, where the stability of structures and reactivity are controlled in the elementary steps (Fig. 1b). The enhanced structural control of chiral Josiphos ligands with different steric or electronic properties may be desirable in separate intermediates and transition states. In the whole cycle, the reaction begins with a stable catalyst in the A conformation, where the Josiphos ligand is located at the metal centre. To overcome the lower activation energy, the conformation of the transition state does not change, resulting in a stable intermediate with A conformation. To improve reactivity and control stereoselectivity, the conformation of the ligand–metal ring is changed to the B conformation via a transition state, leading to the A conformer. Therefore, metal-catalysed asymmetric reactions can be facilitated by a well-designed chiral Josiphos ligand.

The conformational changes of diphosphine ligands and their potential roles in promoting enantioselectivity in transition-metal catalysis.
Combinations of a Pd catalyst and a Josiphos ligand exhibit excellent catalytic performance in asymmetric reactions;22,23,24,25 for example, Pd/Josiphos-catalysed asymmetric amination26 and Pd(TFA)2/Josiphos-catalysed asymmetric Suzuki Miyaura coupling reactions27. The Lin group has reported asymmetric bicyclization/carbonylation of 1,6-enynes employing a Pd catalyst supported by conformationally flexible Josiphos ligands (Scheme 1)28. Three C–C bonds, two rings, two adjacent quaternary carbon stereocenters, and C–O/C–N bonds are formed in this cascade reaction, which exhibits excellent regio- and enantioselectivities. However, several mechanistic questions remain to be addressed: Are two different conformations involved in the same catalytic cycle? Are the reactivity and enantioselectivity determined in the same step or in different steps? How does the ligand affect the reactivity and selectivity in a synergetic fashion? Hence, the mechanism of the Pd-catalysed bicyclization of 1,6-enynes reported by the Lin group is worthy of further study.
In this study, density functional theory (DFT) calculations were used to examine the mechanistic issues mentioned above and to gain insight into how conformational changes affect the entire reaction. Furthermore, we investigated the origin of the enantioselectivity observed in this reaction.

Pd-catalysed cyclopropanation/carbonylation of 1,6-Enynes.
Computational methods
All DFT calculations were performed using the Gaussian 1629 Revision A.03 software package. The B3LYP-D330,31,32,33functional with the standard 6-31G(d) basis set (SDD34,35 basis set for Pd and Fe) was used for geometry optimization in the gas phase. Harmonic vibrational frequency calculations were performed for all the stationary points to determine whether they were local minima or transition structures and to derive thermochemical corrections for the enthalpies and free energies at 298 K. To provide more accurate energy information, the B3LYP-D3 method was used with the 6-311 + G(d) basis set (SDD basis set for Pd and Fe) to calculate the single-point energies in ethyl ether solution. Solvent effects were considered with single-point energy calculations based on gas-phase stationary points with the SMD36,37 continuum solvation model. In the literature, a correction of 1.89 kcal/mol is frequently used to account for the change in standard state from the gas phase (1 atm) to solution (1 M)38,39,40. The independent gradient model (IGM)41 was calculated at the B3LYP-D3/6-31G(d) (SDD for Pd and Fe) level of theory. Multiwfn42 was utilized to produce surface distance projection maps. The geometries of the key reaction intermediates and transition states were drawn using CYLView v1.0 software43.
Results and discussion
As shown in Scheme 2, the general cycle for Pd-catalysed bicyclization/carbonylation of 1,6-enynes begins with Pd(0) catalyst I coordinated to the 1,6-enyne and Josiphos ligand. Oxidative addition of 1,6-enyne with a chiral Pd complex forms the allenylic Pd complex II. Migratory insertion of the olefin group into the C–Pd bond then gives alkyl Pd intermediate III. Subsequent cyclopropanation occurs, releasing one molecule of acetate to afford alkenyl Pd cationic species IV. Two possible pathways were then considered during our computational modelling of this reaction. Carbonylation generates carbonyl Pd species V, which undergoes nucleophilic addition with methanol in the presence of DABCO to generate intermediate VI.

Plausible mechanisms of Pd-catalyzed bicyclization/carbonylation of 1,6-enynes.
Finally, reductive elimination and ligand exchange delivers the 3-azabicyclo[3.1.0]hexane product VII and regenerates the active catalytic species I. An alternative pathway to generate the common intermediate VI involves nucleophilic addition of alkenyl Pd IV followed by carbonylation. With the above proposal in hand, theoretical calculations were performed to reveal the mechanism of the Pd-catalysed bicyclization/ carbonylation of 1,6-enynes. Details of other side reaction mechanisms can be found in the Supporting Information (see Scheme S1, Figure S1 and Figure S2).
Considering the different conformations of Josiphos-coordinated metal, a critical question that remains to be addressed is which conformation is more stable? Furthermore, we calculated the energies of the two conformations of catalyst 1. As shown in Scheme 3, the boat conformation of catalyst 1 is 6.0 kcal/mol lower than that of the half-chair conformer, so it is more stable. Therefore, we choose the catalyst in the stable boat conformation as the catalytic starting point.

Conformations of the ligand-coordinated Pd(0) catalyst 1.
The Josiphos-coordinated Pd(0) catalyst in the boat conformation was chosen as the starting point for the free energy profiles (Fig. 2). Oxidative addition of 1,6-enyne via transition states 2-TS and 2’-TS generates the allenylic Pd(II) complex 3 with the boat conformation, releasing 5.0 kcal/mol free energy. The relative free energy of transition state 2-TS in the boat conformation is 16.1 kcal/mol, which is 2.8 kcal/mol lower than that of the transition state 2’-TS in the half-chair conformation. This result indicates that the transition state of the boat conformation is energetically favourable. Migratory insertion of the olefin group into the C–Pd bond via transition state 6’-TS gives alkyl Pd intermediate 7-R, where the conformation of transition state 6’-TS is the boat conformation. The energy barrier for migratory insertion is 37.0 kcal/mol, which is difficult to overcome. Also, the unstable cationic allenylic Pd(II) complex 3a is generated via transition state 2-TS, wherein the one acetate is dissociated from palladium atom. Then migratory insertion of the alkene via the four-membered cyclic transition state 6a-TS-R with a half-chair conformation occurs to give cationic alkyl palladium intermediate 7a-R with a free energy of 30.2 kcal/mol. The relative energies of the transition states 6a-TS-R is higher than that of 6-TS-R, which is energetically unfavorable (see Figure S3 in the Supporting Information).

Free energy profile of the oxidative addition and migratory insertion steps in the Pd-catalyzed bicyclization/carbonylation of 1,6-enynes. The bond lengths are in Å.
Because of the high energy barrier of the transition state with the boat conformation in the migratory insertion step, an alternative conformation of the transition state was considered. The half-chair conformation of the allenylic Pd(II) intermediate 5 is reversibly obtained by conformational rotation via transition state 4-TS, which is endergonic by 5.8 kcal/mol. At this point, the conformation of the Josiphos-coordinated allenylic Pd(II) complex has changed from a boat conformation to a half-chair conformation. Then, migratory insertion occurs via a half-chair conformational transition state 6-TS-R to generate alkyl Pd intermediate 7-R in the boat conformation. The relative free energy of the half-chair transition state 6-TS-R is 20.0 kcal/mol, which is 12.0 kcal/mol lower than that of the boat conformation transition state 6’-TS. These results indicate that the transition state in the half-chair conformation is more stable than that in the boat conformation, which also confirms our previous speculation that two different conformations are involved in the same catalytic cycle.
To better illustrate the advantages of the half-chair conformation in the migratory step, we used surface distance projection maps and IGM analysis to explore the steric repulsion at different regions of the ligand (Scheme 4). The geometries of Josiphos-Pd are respectively derived from the transition state structures 6-TS-R and 6’-TS-R by omitting the substrates. When using the half-chair conformational isomer, the atoms of Josiphos are far away from the substrate, and the allene group is on the less encumbered side of Josiphos. However, when the boat conformer is used, the ligand atoms are closer to the substrate, resulting in larger steric repulsion. Moreover, IGM analysis revealed the repulsion between the xylyl group of the ligand and the methyl moiety on the allene of the substrate in transition state 6’-TS-R, which would lead to a higher relative free energy. Furthermore, there is a hydrogen bond between the methylene group of the ligand and the acetate in transition state 6-TS-R, which makes the half-chair conformer stable.

a Surface distance projection maps of Josiphos-coordinated Pd. Distances are given in Å. Negative distance (blue) indicates that the atoms on the ligand are farther away from the substrate; positive distance (red) indicates that the atoms on the ligand are closer to the substrate. b IGM analyses of 6-TS-R and 6’-TS-R.
Next, we investigated how the Josiphos-Pd conformers affect enantioselectivity in the migratory insertion step. The migratory insertion transition states leading to the R- and S-enantiomers of the cyclization product are shown in Fig. 3. The transition states in the half-chair conformation (6-TS-R and 6-TS-S) are more stable than those in the boat conformation (6’-TS-R and 6’-TS-S). The energy barrier to generation of the S-enantiomer via 6-TS-S is 27.1 kcal/mol, which is 2.1 kcal/mol higher than that for 6-TS-R, indicating that generation of the S-enantiomer of the cyclization product is unfavourable. This activation energy difference is consistent with the high levels of enantioselectivity observed experimentally. However, when the Josiphos-Pd is in the boat conformation in the migratory insertion step, both transition states are less stable, leading to enantioselectivity flipping (ΔΔG⧧ = −1.6 kcal/mol), which is inconsistent with experimental observations. Therefore, we excluded the transition states in the boat conformation in the migratory insertion step.

Enantioselectivity of the preferred conformers of Josiphos-coordinated Pd(II) in the migratory insertion step.
To further investigate the origin of the enantioselectivity, IGM analysis of the migratory insertion transition states with the half-chair conformation (6-TS-R and 6-TS-S) was performed (Fig. 4). There is a clear steric effect between the methyl group of the allene moiety and the phenyl group of the Josiphos ligand in transition state 6-TS-S. Therefore, the enantioselectivity is mainly controlled by the steric effect in the migratory insertion transition state.
When alkyl Pd intermediate 7-R is generated (Fig. 5), the subsequent dissociation of one acetate from 7-R would provide a cationic Pd center with an empty coordination site. Cyclopropanation via the four-membered cyclic transition state 8-TS in the half-chair conformation occurs to give the alkenyl Pd cationic complex 9 with an energy barrier of 22.4 kcal/mol, and this step is endergonic by 20.5 kcal/mol. The instability of 9 in the boat conformation is contributed to by charge separation and the 14e-configuration. Then, the coordination of carbon monoxide with 9 gives CO-coordinated intermediate 10, which is exergonic by 9.3 kcal/mol. Insertion of carbon monoxide into the Pd–C bond (carbonylation) via the boat conformational transition state 11-TS results in carbonyl Pd intermediate 12 with the half-chair conformation. Subsequently, with the assistance of DABCO, nucleophilic addition of methanol occurs via the half-chair-type transition state 13-TS, resulting in the half-chair conformational intermediate 14 with an energy barrier of 0.6 kcal/mol. Reductive elimination takes place via the half-chair-type transition state.

Enantioselectivity in the migratory insertion step with half-chair ligand conformer and IGM analyses of 6-TS-R and 6-TS-S. The bond lengths are in Å.

Free energy profile of the carbonylation, nucleophilic addition, and reduction elimination steps in Pd-catalysed bicyclization/carbonylation of 1,6-enynes.
15-TS with an energy barrier of 17.1 kcal/mol. The R-enantiomer product is obtained through ligand exchange with 1,6-enyne, regenerating the active catalytic species I.
We also considered the mechanism involving nucleophilic addition followed by carbonylation. It is worth noting that the relative free energy of carbonylation of 20-TS is high at 25.9 kcal/mol, which is 14.8 kcal/mol higher than that of 11-TS, indicating that this pathway is unfavourable.
An alternative cycloaddition route was also explored. As shown in Fig. 6, the Pd(0)-based cyclometallation proceeds through a boat conformational transition state 21-ts with an energy barrier of 68.0 kcal/mol to form palladacyclopentene intermediate 22. In the geometry of 21-ts, the palladacyclopentene species has a distorted five-ring conformation, which may be the origin of its instability. This result excludes the cycloaddition pathway because of its high activation free energy.

Free energy profiles of the cycloaddition step in the Pd-catalyzed bicyclization/carbonylation of 1,6-enynes.
As shown above, the R-enantiomer product is obtained by a catalytic cycle involving oxidative addition, migratory insertion, cyclopropanation, carbonylation, nucleophilic addition, and reductive elimination. In the theoretical calculations, we found that migratory insertion is the rate-determining step, and the enantioselectivity is also controlled in this step. In this transformation, the conformational changes in the Josiphos-Pd are critical to achieving high reactivity and enantioselectivity.
Conclusion
In this work, a computational study on the Pd-catalysed bicyclization/carbonylation of 1,6-enynes indicated that the favourable pathway involves oxidative addition, migratory insertion, cyclopropanation, carbonylation, nucleophilic addition, and reductive elimination. Migratory insertion is both the rate-determining step and the enantioselectivity controlling step. From calculations regarding the mechanism, we discovered that the proper conformation of Josiphos-Pd is essential to achieve the high reactivity and enantioselectivity of this transformation. Dynamic conformational changes allow the catalyst to avoid the high energy barrier and thus improve the reactivity. In migratory insertion transition state, the ligand adopts the less hindered half-chair conformation, which is confirmed by surface distance projection maps and IGM analysis. Throughout the entire catalytic cycle, the intermediates adopt a boat conformation. Furthermore, IGM analysis showed that the steric effect between the phenyl group in the ligand and the methyl group on the allene of the substrate is important for enantioselectivity control. Thus, employing a conformationally self-adaptable ligand is advantageous for enhancing both reactivity and enantioselectivity by regulating ligand-substrate interactions in the key elementary step.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information files.
References
Bochat, A. J., Shoba, V. M. & Takacs, J. M. Ligand-controlled regiodivergent enantioselective rhodium-catalyzed alkene hydroboration. Angew Chem. Int. Ed. 58, 9434–9438 (2019).
Chen, S. et al. Enantioselective construction of six- and seven-membered triorgano-substituted silicon-stereogenic heterocycles. Nat. Commun. 12, 1249–1257 (2021).
Derhamine, S. A. et al. Nickel-catalyzed mono-selective alpha-arylation of acetone with aryl chlorides and phenol derivatives. Angew Chem. Int. Ed. 59, 18948–18953 (2020).
Han, Y. Q. et al. Synthesis of acyclic aliphatic amides with contiguous dtereogenic centers via palladium-catalyzed enantio-, chemo- and diastereoselective methylene C(sp(3))-H arylation. Angew Chem. Int. Ed. 59, 20455–20458 (2020).
Liu, J., Cao, W. B. & You, S. L. Ligand-enabled Z-retentive Tsuji-Trost reaction. Chem. 10, 1295–1305 (2024).
Lu, H. X. et al. Rhodium-catalyzed highly enantioselective hydroboration of acyclic tetrasubstituted alkenes directed by an amide. J. Am. Chem. Soc. 146, 16194–16202 (2024).
Niemeyer, Z. L., Milo, A., Hickey, D. P. & Sigman, M. S. Parameterization of phosphine ligands reveals mechanistic pathways and predicts reaction outcomes. Nat. Chem. 8, 610–617 (2016).
Shan, C., He, M., Luo, X., Li, R. & Zhang, T. Mechanistic insight into anti-carbometalation of an alkyne via η2-vinyl-nickel type Z/E isomerization. Org. Chem. Front. 10, 4243–4249 (2023).
Wang, C., Huang, K., Ye, J. & Duan, W. L. Asymmetric synthesis of P-stereogenic secondary phosphine-boranes by an unsymmetric bisphosphine Pincer-nickel complex. J. Am. Chem. Soc. 143, 5685–5690 (2021).
Kang, T. et al. Multifaceted substrate-ligand interactions promote the copper-catalyzed hydroboration of benzylidenecyclobutanes and related compounds. ACS Catal. 10, 13075–13083 (2020).
Shao, H., Chakrabarty, S., Qi, X., Takacs, J. M. & Liu, P. Ligand conformational flexibility enables enantioselective tertiary C-B bond formation in the phosphonate-directed catalytic asymmetric alkene hydroboration. J. Am. Chem. Soc. 143, 4801–4808 (2021).
Thomas, A. A. et al. Mechanistically guided design of ligands that significantly improve the efficiency of CuH-catalyzed hydroamination reactions. J. Am. Chem. Soc. 140, 13976–13984 (2018).
Zhao, S. et al. Enantiodivergent Pd-catalyzed C-C bond formation enabled through ligand parameterization. Science. 362, 670–674 (2018).
Blaser, H. U., Pugin, B. & Spindler, F. Having Fun (and commercial success) with josiphos and related chiral ferrocene based ligands. Helv. Chim. Acta, 104, e2000192 (2021).
Yasuike, S. et al. Synthesis of JOSIPHOS-type ligands via a diastereoselective three-component reaction and their application in asymmetric rhodium-catalyzed hydroborations. Tetrahedr. Asymm. 16, 3385–3393 (2005).
Zhang, C. Bisphospholane Josiphos-type ligands in rhodium asymmetric catalysis. Chem. Asian J. 18, e202300912 (2023).
Baratta, W., Herdtweck, E., Siega, K., Toniutti, M. & Rigo, P. 2-(Aminomethyl)pyridine – phosphine ruthenium(II) complexes: novel highly active transfer hydrogenation catalysts. Organometallics. 24, 1660–1669 (2005).
Schwenk, R. & Togni, A. P-Trifluoromethyl ligands derived from Josiphos in the Ir-catalysed hydrogenation of 3,4-dihydroisoquinoline hydrochlorides. Dalton Trans. 44, 19566–19575 (2015).
Baidun, M. S., Kalikadien, A. V., Lefort, L. & Pidko, E. A. Impact of model selection and conformational effects on the descriptors for in silico screening campaigns: a case study of Rh-catalyzed acrylate hydrogenation. J. Phys. Chem. C. 128, 7987–7998 (2024).
Gallagher, J. M., Roberts, B. M. W., Borsley, S. & Leigh, D. A. Conformational selection accelerates catalysis by an organocatalytic molecular motor. Chem. 10, 855–866 (2024).
Peng, Q., Wang, Z., Zaric, S. D., Brothers, E. N. & Hall, M. B. Unraveling the role of a flexible tetradentate ligand in the aerobic oxidative Carbon-Carbon Bond formation with Palladium complexes: a computational mechanistic study. J. Am. Chem. Soc. 140, 3929–3939 (2018).
Chen, Y. W., Liu, Y., Lu, H. Y., Lin, G. Q. & He, Z. T. Palladium-catalyzed regio- and enantioselective migratory allylic C(sp(3))-H functionalization. Nat. Commun. 12, 5626–5634 (2021).
Maitro, G. et al. Enantioselective synthesis of aryl sulfoxides via palladium-catalyzed arylation of sulfenate anions. Org. Lett. 9, 5493–5496 (2007).
Shen, Q., Shekhar, S., Stambuli, J. P. & Hartwig, J. F. Highly reactive, general, and long-lived catalysts for coupling heteroaryl and aryl chlorides with primary nitrogen nucleophiles. Angew Chem. Int. Ed. 44, 1371–1375 (2005).
Xu, Q. & Han, L. B. Palladium-catalyzed asymmetric hydrophosphorylation of norbornenes. Org. Lett. 8, 2099–2101 (2006).
Dolja, E., Funken, N., Slak, D., Schnakenburg, G. & Gansäuer, A. A divergent duo: palladium catalyzed carboamination in enantioselective desymmetrization and regiodivergent catalysis. ChemCatChem. 11, 5421–5424 (2019).
Kim, J. & Cho, S. H. Access to enantioenriched benzylic 1,1-silylboronate esters by palladium-catalyzed enantiotopic-group selective Suzuki–Miyaura coupling of (diborylmethyl)silanes with aryl iodides. ACS Catal. 9, 230–235 (2018).
Li, Q. et al. Pd-catalyzed asymmetric 5-exo-trig cyclization/cyclopropanation/carbonylation of 1,6-enynes for the construction of chiral 3-azabicyclo[3.1.0]hexanes. Angew Chem. Int. Ed. 62, e202211988 (2023).
Frisch, M. J., et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, C., 2016. The full author list is shown in the Supporting Information.
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Becke, A. D. & Johnson, E. R. A density-functional model of the dispersion interaction. J. Chem. Phys. 123, 154101–154110 (2005).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104–154123 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Dolg, M., Stoll, H. & Preuss, H. Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 90, 1730–1734 (1989).
Dolg, M., Wedig, U., Stoll, H. & Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 86, 866–872 (1987).
Cancès, E., Mennucci, B. & Tomasi, J. A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 107, 3032–3041 (1997).
Cossi, M., Barone, V., Cammi, R. & Tomasi, J. Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 255, 327–335 (1996).
Bryantsev, V. S., Diallo, M. S. & Goddard, III. Calculation of solvation free energies of charged solutes using mixed cluster/continuum models. J. Phys. Chem. B. 112, 9709–9719 (2008).
Kelly, C. P., Cramer, C. J. & Truhlar, D. G. SM6: a density functional theory continuum solvation model for calculating aqueous solvation free energies of neutrals, ions, and solute-water clusters. J. Chem. Theory Comput. 1, 1133–1152 (2005).
Kelly, C. P., Cramer, C. J. & Truhlar, D. G. Aqueous solvation free energies of ions and ion-water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton. J. Phys. Chem. B. 110, 16066–16081 (2006).
Lefebvre, C. et al. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 19, 17928–17936 (2017).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Legault, C. Y. C., 1.0b, Université de Sherbrooke, (2009). https://www.cylview.org).
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (Grant No. 22103008 and 21903071), the China Postdoctoral Science Foundation (2022M710548), Scientific and Technological Research Program of Chongqing Municipal Education Commission (KJQN202400524), Scientific Research Foundation of Chongqing Normal University (21XLB013).
Author information
Authors and Affiliations
Contributions
Conceptualization: CS, XL, YL; Writing—original draft: XL, CS; Supervision: CS, YL; Project administration: CS, YL; Funding acquisition: CS, YL. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shan, C., Liu, X., Luo, X. et al. Theoretical study on ligand conformational self-adaptation for modulating reactivity. Sci Rep 14, 24031 (2024). https://doi.org/10.1038/s41598-024-75141-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-75141-6
