
Abstract
Recent research highlights the significant impact of the gut microbiota on health and disease. Thalassemia, a hereditary blood disorder, requires regular blood transfusions, leading to an accumulation of iron in the body. Such changes could potentially alter the intestinal microbiota, thereby increasing the susceptibility of thalassemic patients to infection. In this study, we analyzed the fecal microbiota of 70 non-transfusion-dependent (NTDT) β-thalassemia/HbE patients and 30 healthy controls. Our findings indicate that iron chelation intervention had no detectable effect on the microbiome profile of thalassemic patients. However, the cross-sectional analysis revealed that the bacterial diversity and community structure in patients were significantly less diverse and distinct compared to those of healthy subjects. Using reference frames, we were also able to demonstrate that bacterial taxa that are known to produce short chain fatty acids, from the genera Alistipes, Coprococcus, and Oscillospira, and those from the family Ruminococcaceae, were less prevalent in the patients. In contrast, bacterial taxa associated with an unhealthy gut, including the genus Clostridium and those from the families Fusobacteriaceae, Enterobacteriaceae, and Peptostrptococcaceae, were more prevalent in patients and found to be correlated with higher levels of ferritin. Collectively, these changes in the microbiota could be regarded as markers of raised ferritin levels, and therefore, awareness should be exercised as they could interfere, albeit indirectly, with the treatment of the co-morbidities of thalassemia.
Similar content being viewed by others


Introduction
Thalassemia is a hereditary blood disorder caused by an excess of unpaired globin chains due to defective globin synthesis. Point mutations in the β-globin gene lead to the absence of or reduced production of β-globin, giving rise to β-thalassemia, in which the imbalance between the β- and α-globin chains results in ineffective erythropoiesis and, consequently, hemolysis1. Hemolytic anemia, splenomegaly, and iron overload are well-known characteristics of β-thalassemia. Severe hemolytic anemia necessitates frequent blood transfusions, a common requirement in thalassemia management2,3. Transfused blood contains iron, which is stored in the tissues by binding to ferritin, a protein that stores iron4,5,6. When iron content exceeds the capacity of ferritin, this free iron, together with products of free α-globin chain breakdown, catalyzes the formation of free radicles, causing a multitude of organ and cellular damage7. Endocrine, hepatic, and cardiac tissues are the most affected by iron deposition resulting in a serious condition associated with morbidities and, in many instances, mortality8,9,10. Therefore, iron chelation therapy plays a crucial role in contributing to the significant improvement in the survival of thalassemic patients. This therapeutic approach, typically involving iron chelators such as desferrioxamine, deferiprone, and deferasirox, is initiated after 10–20 transfusions or when serum ferritin levels exceed 1000 µg/L to prevent adverse effects of iron overload3,9.
Excess iron not only directly cause catastrophic effect at cellular and organs level but the iron is known to have effects on the gut microbiota composition11,12,13,14. An altered intestinal microbiota has been shown to be associated with chronic conditions such as obesity, irritable bowel disease, atherosclerosis, liver diseases, and liver cancer, as well as genetic disorders such as autism spectrum disorder15,16,17. In particular, iron can disrupt the balance towards the growth of infectious organisms, as shown in previous studies where iron fortification affected the balance of intestinal commensal bacteria by favoring the growth of pathogenic bacteria18,19. Moreover, studies in iron-overloaded thalassemia mice showed that the gut mucosal injury releases lipopolysaccharides, from Gram-negative bacteria residing in the gut, which themselves are a cause of inflammatory responses20 and enhanced sepsis21. A recent study on mice showed that both intravenous infusions and blood transfusions can cause iron overload, which raises the amount of iron in the feces and changes the gut microbiota in a way similar to dietary iron22. Thus, it is unsurprising that infections and sepsis are the major causes of morbidity and mortality in β-thalassemia23,24.
Gut inflammation and gut barrier disruption are not solely influenced by changes in the composition of gut microbiota but also by alterations in the metabolites produced by these altered microbial communities25. Short-chain fatty acids (SCFAs), the gut microbiota-producing metabolites, play a crucial role in preserving gut health by promoting the integrity of the intestinal barrier, therefore inhibiting the colonization of pathogenic microbes26,27. Additionally, SCFAs possess potent anti-inflammatory characteristics by regulating immunological responses and suppression of pro-inflammatory cytokine synthesis28. These roles are crucial in β-thalassemia, where excessive iron intake from recurrent blood transfusions worsens inflammation of the stomach and degeneration of the epithelium29,30.
These incidental studies raise an interesting question as to whether iron overloading among thalassemic patients, resulting from increased iron absorption and frequent blood transfusions, could alter the intestinal microbiome, especially those that are involved in SCFA production, and consequently explain the high rate of susceptibility to infection in these patients. In our study, we analyzed changes in the gut microbiota using fecal samples of thalassemic patients and observed significant alterations in the bacterial diversity and community structure of the gut microbiota. We also found that some bacterial taxa that are known to promote or prevent infections are significantly altered in thalassemic patients. This study also presented a correlation between iron status and unhealthy gut-associated bacteria taxa. This discovery of dysbiosis in thalassemic patients is one of the first studies on the microbiome composition in thalassemia and provides some possible insight on the prognosis and treatment outcomes of infections in thalassemic patients.
Results
Participant and characteristics
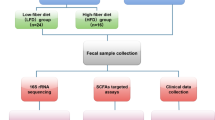
The study was conducted in Nakhon Pathom province, Thailand. We recruited 70 non-transfusion-dependent (NTDT) β-thalassemia/HbE patients and 30 healthy subjects. Detailed inclusion and exclusion criteria can be found in Supplementary Table 1. This study was approved by the Mahidol University ethics committee (Approved protocol number MU-CIRB 2018/181.1309 and COA No. MU-CIRB 2018/184.2610), and written informed consent was obtained from all participants. The patients were classified into three groups of iron overload based on their serum ferritin level: Low (< 1,000 ng/ml, n = 39), Medium (1000–2000 ng/ml, n = 15) and High (> 2000 ng/ml, n = 16). The healthy subjects were recruited from people living in the same environment as the patients (n = 30). The major characteristics of the cohort can be found in Table 1.
Iron chelation intervention shows no detectable longitudinal effect on microbiome profile of thalassemic patients in this study
One of the questions we aimed to address in this study was whether iron chelator administration has a detectable effect on a patient’s microbiome profile. Thus, we designed and conducted a longitudinal study focusing on the effect of iron chelation intervention by comparing patients’ microbiota profiles before (Stop period, T1-T3) and after (Resume period, T4-T5) iron chelation (Fig. 1). We determined the Shannon diversity index to test if the alpha diversity of patients’ microbiota between the two time periods (Stop vs. Resume) was significantly altered compared to healthy subjects. However, no significant difference in the Shannon diversity index was observed between Stop and Resume periods in all patient groups (Low, Medium, and High ferritin) when compared to the changes in the Shannon diversity index observed in the healthy subjects during the same period (Supplementary Fig. 1a, b). Also, beta diversity based on unweighted-UniFrac displayed that no significant difference was observed between Stop and Resume periods among all patient groups (Supplementary Fig. 1c, d). Altogether, these findings suggest that the effect of iron chelators on the microbiota composition of patients could not be detected in this study. Therefore, the remainder of the study will focus solely on a cross-sectional study of the microbiome profiles of thalassemia patients and healthy subjects prior to the administration of iron chelators (Stop period, T1-T3).

Study design. Initial screening involved a baseline health assessment, including measurements of ferritin and hemoglobin levels, conducted 14 days prior (day − 14). Following enrollment, stool samples were collected at five timepoints to assess changes in the microbiome due to iron chelation therapy. The first three samples (T1-T3) were taken on days 0, 5, and 7. After resuming iron chelation on day 7, the final two samples (T4 and T5) were collected on days 9 and 14.
The microbiome profiles of thalassemic patients are distinct and less diverse than those of healthy subjects
We first investigated whether the microbiota of thalassemic patients differed from that of healthy subjects. From all 100 enrolled participants, a total of 300 samples were collected during the Stop period (T1, T2, and T3 timepoints). After data processing, the total number of qualified samples was reduced to 292 and subsequently used for the rest of the analysis in the study. The Shannon index (Fig. 2a) and Faith’s phylogenetic diversity (Fig. 2b) in healthy subjects (4.307 and 11.678, respectively) were significantly higher than those in thalassemic patients (3.937 and 9.453, respectively), suggesting that thalassemic patients have less microbial diversity than healthy subjects. Next, compositional Aitchison β diversity of samples was analyzed to elucidate the nature of community differences between thalassemic patients and healthy subjects. Non-metric multidimensional scaling (NMDS) analysis suggests that bacterial communities among two groups were significantly different (p = 0.001, PERMANOVA), especially along the NMDS 1 axis (Fig. 2c).

Bacterial community of thalassemia patients are less diverse and distinct compared to healthy subjects’ microbiome profile. Box plots displaying (a) Shannon index and (b) Faith’s phylogenetic diversity index in two different groups. These values were compared to others in a pairwise manner with the Kruskal-Wallis test (α = 0.05). (c) Non-metric multidimensional scaling (NMDS) biplot based on Aitchison distance. MetaMDS and envfit functions (vegan package) in R were calculated and used to display correlations between microbial community compositions and environment metadata. Dots represent individual samples, where blue color represents normal and red color represents disease group. Vectors (arrows) represent the direction of metadata in relation to normal or disease groups. The comparison between two groups (Normal vs. Disease) of box plots was calculated based on the Wilcoxon test.
Due to the notable differences in diversity observed, we looked into what factors caused the differences in the bacterial communities between thalassemic patients and healthy subjects. While there was no significant difference in age between the patients and healthy subjects (p > 0.1, t-test), BMI (p < 0.001), ferritin level (p < 0.001), and hemoglobin level at screening (p < 0.001) were significantly different (Table 1). BMI was significantly higher in healthy subjects compared to the thalassemic patients, which is in agreement with previous studies demonstrating that the BMI of the Thai population (young men) ranges from 21.7 kg/m2 to 22.5 kg/m231, while thalassemic patients normally have a lower BMI32. Next, correlations between microbial community composition and environmental metadata were analyzed (Fig. 2c). The results show that body mass index, weight, and HB levels at screening were negatively correlated with bacterial community composition (envfit; body mass index: R2 = 0.0923, p-value = 0.001, weight: R2 = 0.0455, p-value = 0.002, HB level: R2 = 0.1750, p-value = 0.001), while ferritin level, height, and age were positively correlated (envfit; ferritin level: R2 = 0.0783, p-value = 0.001, height: R2 = 0.0354, p-value = 0.008, and age: R2 = 0.0366, p-value = 0.005). Notably, the HB level at screening and the ferritin levels had a strong effect on the make-up of the microbial communities in healthy people and thalassemic patients.
Higher level of Proteobacteria and Fusobacteria phylum observed in thalassemic patients
Since changes at the phylum level have been used as indicators of various dysbiosis15,33, we next investigated changes in microbial population at the phylum level in both groups. At the phyla level (Fig. 3a), Bacteroidetes was the dominant phylum in both groups, with an average of 44.03%, followed by Firmicutes at 33.33% on average. Proteobacteria, Fusobacteria, Actinobacteria, and Verrucomicrobia were at 16.87%, 0.02%, 0.02%, and 0.01%, respectively. Significant differences in the relative abundance of phyla could be seen between the normal and diseased groups (Fig. 3a). Proteobacteria were higher in the disease group at 9%, whereas 5.59% in the normal group.

Changes in microbial population at phylum level in the normal and thalassemia groups. (a) A boxplot displaying the relative abundance (%) of five main phyla in normal (blue) and disease (red) groups. A Wilcoxon two-sided test was used to calculate the significance (p-value < 0.05). (b) Boxplots showing the log-ratios of phyla Proteobacteria and Fusobacteria to those of the phylum Bacteroidetes. All log–ratios are statistically significant, based on a two-sample t-test (*** p < 0.001).
Similarly, Fusobacteria exhibited higher abundance in the diseased group at 0.65%. while it was absent (0%) in the normal group. The differences in the relative abundances of both Proteobacteria and Fusobacteria between the diseased and normal groups were statistically significant (Wilcoxon two–sided test; P–value < 0.05). On the other hand, differences in the relative abundance between normal and diseased groups were also observed for other phyla but were not statistically significant. This was the case for the relative abundances of Bacteroidetes, at 52.44% and 44.90%, Firmicutes at 29.49% and 28.1%, and Actinobacteria at 0.63% and 0.64%, for the normal and diseased groups, respectively. Due to the compositional effect of these sequencing datasets, Songbird and log-ratios are preferred to determine the differences between the groups34. Since Bacteroidetes is one of the two most abundant phyla in the human gut microbiome35 and also showed no significant difference between normal and thalassemic groups in our study, it was used to calculate log-ratios as a reference frame for other phyla. In agreement with the phylum relative abundance data (Fig. 3a), the log-ratios of the phyla Proteobacteria and Fusobacteria to the phylum Bacteroidetes were significantly higher in the disease group compared to the healthy group, indicating that both phyla are prevalent in thalassemic patients (Fig. 3b).
Short-chain fatty acids (SCFA) producing bacteria are prevalent in healthy subjects, while unhealthy gut-associated bacteria are prevalent in thalassemic patients
Identifying taxonomic signatures in microbiome datasets that can infer the health status of the subject is crucial for disease marker development. In this study, we used Songbird34 and Qurro36 to find the differential ranks of each taxa, determining which taxa are prevalent in healthy subjects and which in thalassemic patients. The analysis revealed that taxa that are known to be associated with an unhealthy gut, including the genus Clostridium, the families Fusobacteriaceae, Enterobacteriaceae, and Peptostrptococcaceae, are prevalent in thalassemic patients. In contrast, taxa associated with SCFA production, such as the genus Alistipes37, Osillospira38, Coprococcus39, and the family Ruminococcaceae40, are more prevalent in the normal group (Fig. 4a). The full set of differential rank is listed in Supplementary Table S2. Since the genus Bacteroides was prevalent and most abundant in both groups according to the differential ranking and also showed no significant difference between the normal and disease groups (Supplementary Fig. 2), it was used as the reference frame to calculate log-ratios for other taxa. Indeed, the log-ratios of unhealthy gut-associated taxa (Fig. 4b) to the genus Bacteroides were significantly higher in the disease group compared to the healthy group. In contrast, log-ratios of SCFA-producing taxa were significantly lower in the disease group (Fig. 4c). Together, these findings suggest that, in healthy individuals, bacteria that produce SCFAs are common, whereas thalassemic patients tend to have a higher prevalence of unhealthy gut-associated bacteria.

Unhealthy gut-associated, but not SCFA-producing bacteria are more prevalent in thalassemic patients. (a) Differential rankings of taxa associated with disease status determined by Songbird and illustrated in Qurro. Each taxon is represented by different color bars in the plot. (b) Boxplots showing log-ratios of different unhealthy gut-associated bacterial taxa to that of genus Bacteroides. (c) Boxplots showing log-ratios of different SCFA-producing bacterial taxa to those of the genus Bacteroides. All log-ratios are statistically significant, based on a two-sample t-test (*p < 0.05, ** p < 0.01 and *** p < 0.001).
Higher ferritin levels are linked to unhealthy gut-associated bacteria abundance
Given that iron-overloaded conditions are commonly observed in thalassemic patients, the increase in the unhealthy gut-associated bacteria found in patients urged us to investigate if elevated iron levels, as indicated by ferritin levels, are associated with the abundance of these bacteria. We investigated correlations between microbiota relative abundance and hematological parameters, ferritin level and HB levels, in all 292 samples, including both healthy subjects and thalassemic patients, by employing Spearman’s correlation test (Fig. 5). The results showed that ferritin level was positively correlated with the relative abundance of certain unhealthy gut-associated bacterial taxa: Clostridium genus, Enterobacteriaceae family, Fusobacteriaceae family, and Peptostreptococcaceae family (Fig. 5a and b). Conversely, bacteria known for SCFA production, i.e., Prevotella genus41, Lachnospira genus42, Roseburia genus43, Alistipes genus37, Osillospira genus38, Coprococcus genus39 and Ruminococcaceae family40, showed a negative correlation with the ferritin levels (Fig. 5a). In thalassemia, ferritin levels are known to inversely correlate with HB levels due to the pathogenicity of the disease7. Thus, it is unsurprising that the relationship between bacteria abundance and ferritin level is opposed to the bacteria abundance and HB levels. Together, these results suggest that ferritin levels are correlated with unhealthy gut-associated bacteria, while HB levels are correlated with SCFA-producing bacteria in participants.

Higher ferritin level is linked to higher abundance of unhealthy gut-associated bacteria (a) Spearman correlations, visualized by the MicrobiomeSeq and phyloSeq packages in R, showing relative abundance of bacteria correlating with ferritin and HB (hemoglobin) levels. The adjusted p-values were calculated using Benjamini and Hochberg (***p = 0.001, **p = 0.01, *p = 0.05). (b) Graphs showing relative abundances (%) of unhealthy gut-associated bacteria are significantly correlated with high ferritin levels.
Discussion
NTDT β-thalassemia/HbE patients were specifically selected for this study due to their predominance in the patient population registered at our designated clinic. Although NTDT patients require fewer blood transfusions, they still experience a certain degree of iron overload. Furthermore, the reduced need for blood transfusions in NTDT patients is associated with a more stable clinical profile compared to those with transfusion-dependent thalassemia (TDT). This stability during the course of our investigation allowed us to minimize confounding variables related to frequent transfusions and their associated complications, thereby enabling a more accurate assessment of the microbiome alterations. Given the potential differences in the gut microbiome between NTDT and TDT patients, it is recommended that future studies focusing on TDT patients would be crucial for dissecting the specific effects of chronic iron overload and frequent transfusions on the gut microbiome in TDT patients.
In this study, we first found that the effect of iron chelators on the microbiota composition of patients could not be detected in the longitudinal study. Consistent with our findings, previous research conducted in a murine model demonstrated that treatment with deferiprone, an iron chelator, failed to alleviate gut dysbiosis in heterozygous β-thalassemia mice44. This can be partially attributed to the fact that iron chelators do not have direct bactericidal effects45. Therefore, it is not anticipated that there will be sudden alterations in the microbiome profile following the administration of an iron chelator, as would often occur after taking antibiotics46. Moreover, long-term treatment is recommended to achieve and maintain reduced iron levels in organs and tissues, for example, thalassemic patients receiving iron chelator therapy are required to have their liver iron concentrations monitored every 6 months and serum ferritin every 3 months, in order to observe a change in iron levels47. Therefore, the length of the iron chelation intervention in this study might not have been adequate to detect changes in the microbiome profile of the patients. Furthermore, the relatively low number of patients in each group (Low, Medium, and High ferritin) might undermine the analysis power of the study, resulting in the undetectable changes in diversity indexes caused by iron chelation intervention. It is well-documented that determining the minimum number of participants required to find statistically significant differences in clinical microbiome studies is a largely unresolved subject, especially when the preliminary data of the microbial diversity in the targeted population is unavailable48. Given the lack of prior information on the microbial diversity of the NTDT-thalassemic patients, the sample size for this study was primarily determined based on feasibility considerations. Despite this limitation, this study provides valuable baseline data on the microbial diversity of NTDT-thalassemic patients for future research investigating microbiome changes that are clinically relevant to interventions such as iron chelation therapy.
A cross-sectional study of the microbiome profile demonstrated that the gut microbiota in fecal samples from thalassemic patients exhibited lower diversity and distinctiveness compared to those from healthy subjects. Contrary to a previous study on thalassemic mouse models where iron overload did not affect the diversity of the fecal microbiome10, our findings align with studies on humans with various chronic conditions such as diabetes49, allergic asthma50, liver cirrhosis51, and inflammatory bowel disease (IBD)52. These studies also observed less diverse and distinct microbiota profiles among diseased groups. At the phylum level, we observed that Proteobacteria and Fusobacteria were significantly higher in thalassemic patients. Similar to our findings, the higher abundance of Proteobacteria has been found to be associated with intestinal inflammation11, colitis, and IBD53. Fusobacteria has also been found to be enriched in immunocompromised patients54 and patients suffering from liver cirrhosis51.
Regarding the changes in SCFA-producing bacteria, in this study, we observed a reduced prevalence of bacterial taxa from genera such as Alistipes, Coprococcus, and Oscillospira, and those from the family Ruminococcaceae, all of which are SCFA-producing bacteria37,38,39,40. The low prevalence of SCFA-producing bacteria in thalassemic patients may result in decreased SCFA production, leading to compromised intestinal immunity and diminished ability to maintain the integrity of intestinal epithelial cells55. Particularly, bacteria from the family Ruminococcaceae was reported to be significantly reduced in patients with IBD40, and species from the genus Alistipes have been shown to possess anti-inflammatory and tumor-suppressive properties, protecting the gut against colitis37. Both Ruminococcaceae and Alistipes were less prevalent in thalassemic patients in our study, and therefore, it is not surprising that thalassemic patients are prone to systemic infections with sepsis and liver abscess, among others, being common23,24,56. Additionally, as high levels of SCFAs have been shown to inhibit the growth of Enterobacteriaceae57,58, this could possibly explain the higher prevalence of Enterobacteriaceae in thalassemic patients. Blooms of Enterobacteriaceae have been known to enhance the susceptibility of the gut to pathogens such as Clostridium difficile57, which also coincides with our finding of a higher prevalence of the Clostridium genus thalassemic patients. On the other hand, Lachnospiraceae, which was known to hinder Clostridium difficile colonization59, was less prevalent in thalassemic patients in our study. Whether the bacterial taxa and their members identified in this study play significant roles in clinical outcomes, such as increased susceptibility to intestinal infections, could be an intriguing topic for future studies.
Besides, healthy subjects in many studies were shown to have a high abundance of Lachnospira, Alistipes, and Roseburia, suggesting their association with good health37,42,60. Our data showed similar results, as these genera had positive correlations with hemoglobin levels, indicating a healthy state of participants. A previous study reported that Oscillospira was also found to be closely related to healthy humans and that it was positively correlated with a diverse microbial environment61. Moreover, negative correlations have been found between Oscillospira and Clostridium, with the latter being able to produce metabolites that could render an unfavorable environment for the growth of Oscillospira. This report supports our findings that the relative abundance of Clostridium correlated with ferritin levels, indicating disease, whereas that of Oscillospira correlated with hemoglobin levels, indicating normal health. Additionally, we observed that Copprococcus, a Gram-positive coccus regarded as an indicator of high quality of life, was correlated with hemoglobin levels.
Studies in both adults and children have shown that fecal Enterobacteriaceae are correlated with increased levels of serum ferritin11. Enterobacteriaceae can, in turn, enhance the growth of disease-associated bacteria, particularly Clostridium difficile57. These findings are consistent with our results, as we observed a correlation between increased ferritin levels and the presence of the genus Clostridium and families Enterobacteriaceae. It is worth noting that, in this study, only ferritin level was used as an indicator of the systematic iron status of the participants. Thus, further studies are needed to dissect whether the prevalence of unhealthy gut-associated bacteria in gut microbiome of thalassemic patients is associated with the fecal iron level similar to the ferritin levels shown in this study.
Other limitations of the study should also be acknowledged. Firstly, it is necessary to take into account the variability in the collection and storage of fecal samples. Although previous research has demonstrated that interpersonal variations in gut microbiota profiles generally outweigh changes in microbial populations as a result of storage conditions62, it is widely acknowledged that the methodology used in microbiome analysis can have a significant impact on study outcomes63,64,65. Consistent protocols were implemented for all samples in this study, including DNA extraction, sequencing technologies, transit conditions, and storage temperature. However, the precise duration of sample storage prior to shipment could not be uniformly controlled. Research has shown that the composition of the microbiome can be influenced by differences in storage timing to varying degrees, depending on the study design66,67. As well as the variation resulting from sample storage, other participant-related factors may also contribute to the observed variations in microbiome profiles. We were unable to rigorously regulate the time of day during which patients visited the clinic and collected their samples on-site. The microbiome composition may be influenced by the timing of sample collection, as suggested by prior research68,69. In addition, the dietary intake of participants was not standardized, which is likely to have contributed to the individual variations in microbiome profiles. Diet is a well-established factor that influences the human microbiome, as studies have shown that dietary changes can swiftly alter the microbiome composition within days70,71,72. Consequently, although this study identified differential microbiome profiles and specific dominant species that are associated with iron overload in thalassemic patients, it is crucial to take into account the potential covert effects of sample handling and storage as well as the absence of control over the food consumption of participants during the study period when interpreting the microbiome shifts that were observed in this study.
The potential clinical applications of microbiome profiling in the prevention and treatment of diseases are becoming more evident, paving a way for personalized therapeutic interventions73,74. For example, the involvement of the gut-liver axis in liver injury caused by excess iron was highlighted, indicating that targeting specific gut microbes could alleviate liver damage73. Furthermore, clinical applications could be extended to monitoring of microbiome profiles as indicators of disease progress, particularly in the management of sepsis, where the gut microbiome has been recognized as a promising therapeutic target74,75. In particular, profiling the microbiota in β-thalassemia patients, where excessive iron intake compromises gut integrity, could contribute to the identification of individuals with an increased susceptibility to severe mucositis and sepsis20,21. Our results indicate that thalassemic patients have a higher prevalence of pathogenic bacteria, which are also associated with elevated ferritin levels. Therefore, it is crucial to regard these bacteria as indicators of increased ferritin levels. When the microbiome of thalassemic patients exhibits an abundance of these bacteria, caution is warranted, as they may indirectly impact the outcomes of treating thalassemia co-morbidities. In order to restore healthy gut microbiota and prevent pathogen translocation that leads to systemic infections, future research should prioritize the development of microbiome-based diagnostics and therapeutics, such as phage therapy, probiotics, or prebiotics76,77,78.
Materials and methods
Study design and sample collection
For the primary screening process, all participants were reviewed at baseline (-14 day, Fig. 1) for health assessment, measuring of ferritin and hemoglobin levels according to a previous study79. Once enrolled, all thalassemic patients were asked to discontinue iron chelation for 14 days before the first sample collection. Stool samples were collected at five different timepoints to evaluate longitudinal microbiome alteration caused by iron chelation therapy intervention. Stool samples from the first three time points (T1-T3) were collected on days 0, 5, and 7, respectively. After day 7, iron chelation therapy was resumed for patients. Stool samples for the last two time points, T4 and T5, were collected at days 9 and 14, respectively. For healthy subjects, all five time points were collected within the same time interval as those of the patients, but without any intervention. The experimental design is summarized in Fig. 1. Stool collection procedures were carried out using BD BBL™ CultureSwab™ EZ II. Once collected, samples were kept at 4 °C during transport and frozen at − 20 °C within 1 h. Since recruitment spanned over the course of 12 months, some samples were kept frozen longer than the others. However, this was consistent across all groups for comparison purposes.
DNA extraction
Procedure for the extraction of genomic DNA from fecal samples were modified from MagAttract® PowerSoilⓇ DNA KF Kit (384) (QIAGENⓇ catalogue: 27000-4-KF). The sample swabs were processed in each well of a PowerBead DNA plate followed by the robotic program on the KingFisher Flex Purification System80.
16 S sequencing
An amplicon library was constructed, using the previously extracted genomic DNA, by amplifying the V4 region of 16S ribosomal RNA gene. The primers used for this amplification were FWD 515F 5’ – GTGYCAGCMGCCGCGGTAA – 3’ and REV 806R 5’ – GGACTACNVGGGTWTCTAAT – 3’ (https://earthmicrobiome.org/protocols-and-standards/16s/). PCR reaction was carried out on a thermocycler, and the amplicon obtained was quantified and sequenced on Illumina Platform, in accordance with the previous protocol (https://doi.org/10.17504/protocols.io.nuudeww) and the previous study81.
HB level and ferritin level assessment
Three mL of an EDTA blood tube were used for hematological and Hb analyses using the automatic HPLC (Bio Rad, Variant II system), while 4 mL were used for serum ferritin measurement using immunoenzymatic kits (DiaMetra, Segrate, Italy).
Bioinformatic analysis
Raw sequencing data in FASTQ format were processed using QIIME 2 (version 2019.10)82 based on Deblur83, which provides single-nucleotide resolution based on error profiles within samples. First, paired-end FASTQ reads from each sample were merged using vsearch84 to join paired-end reads. Next, merged reads were filtered based on read quality using QIIME 2’s built-in quality-filter q-score-joined script. Then, filtered reads were processed with Deblur, using a trim length of 250 base pairs to obtain amplicon sequence variants (ASV). A phylogenetic tree was then generated for phylogenetic diversity analyses. A phylogenetic tree was built using the q2-fragment-insertion plugin85 based on Greengenes 99% identity database. The amplicon sequence variants (ASVs) were subsampled, based on the lowest count sequences (rarefied to 2000 to minimize the effects of sequencing depth) for alpha and beta analysis using the core-metrics-phylogenetic method.
Alpha diversity metrics were used to estimate microbial diversity based on Shannon and Faith’s phylogenetic diversity between two groups. Pairwise comparisons of alpha diversity were calculated using a Kruskal-Wallis test. Moreover, the Shannon diversity index was also estimated to test whether alpha diversity changed significantly between two different time periods (stop and resume) according to ferritin level with q2-longitudinal plugin86.
Beta diversity analysis, including robust Aitchison and unweighted-UniFrac distance, was calculated using the beta-group-significance command in QIIME 2. Permutational multivariate analysis of variance (PERMANOVA) based on unweighted-UniFrac was also used to analyze statistical differences in beta diversity with QIIME 2. Moreover, a non-metric multidimensional scaling (NMDS) plot, displayed as a biplot, showed correlations between microbial community composition and environment metadata. NMDS based on Aitchison distance with biplot, was calculated by metaMDS and envfit functions (vegan package) in R87. A pairwise comparison of beta diversity based on Aitchison distance was calculated using the Wilcoxon test. For taxonomy analysis, it was assigned to ASVs using Greengenes (version 13_8) database classifier. A non-parametric two-group comparison was performed with the Wilcoxon test.
The differential rankings of taxa associated with thalassemia and the normal group were determined using Songbird34. ASVs generated by QIIME 2 were used as input for Songbird to rank taxa associations with the different groups, with normal samples serving as a reference. Qurro36 was also used to calculate and visualize the differential ranks of taxa associated with thalassemia versus the normal group. Moreover, correlations between the relative abundance of genus and ferritin level were calculated based on Spearman’s correlation, and p-value were adjusted using the Benjamini and Hochberg method88. The correlation coefficients were visualized using MicrobiomeSeq (https://github.com/umerijaz/microbiomeSeq.git) and the phyloSeq package in R89.
Data availability
The data presented in this study are available in the manuscript and supplementary materials. Other data are available on request from the corresponding author. Some data is not publicly available due to the Personal Data Protection Regulation.
References
Fucharoen, S. & Winichagoon, P. New updating into hemoglobinopathies. Int. J. Lab. Hematol.34, 559–565 (2012).
Musallam, K. M., Rivella, S., Vichinsky, E. & Rachmilewitz, E. A. Non-transfusion-dependent thalassemias. Haematologica. 98, 833–844 (2013).
Rachmilewitz, E. A. & Giardina, P. J. How I treat Thalassemia. Blood. 118, 3479–3488 (2011).
Taher, A., Vichinsky, E., Musallam, K., Cappellini, M. D. & Viprakasit, V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT) (Thalassaemia International Federation, 2013).
Gardenghi, S. et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 109, 5027–5035 (2007).
Lithanatudom, P. et al. A mechanism of ineffective erythropoiesis in β-thalassemia/Hb E disease. Haematologica. 95, 716–723 (2010).
Nadia Maria Sposi. Oxidative Stress and Iron Overload in β-Thalassemia: An Overview. in Beta Thalassemia (eds. Marwa Zakaria & Tamer Hassan) Ch. 6. doi:IntechOpen, Rijeka, (2019). https://doi.org/10.5772/intechopen.90492
Taher, A. T. & Saliba, A. N. Iron overload in Thalassemia: different organs at different rates. Hematology. 2017, 265–271 (2017).
Taher, A., Isma’eel, H. & Cappellini, M. D. Thalassemia intermedia: revisited. Blood Cells Mol. Dis.37, 12–20 (2006).
Visitchanakun, P. et al. Gut leakage enhances sepsis susceptibility in iron-overloaded β-thalassemia mice through macrophage hyperinflammatory responses. Am. J. Physiol. - Gastrointest. Liver Physiol.318, G966–G979 (2020).
Kortman, G. A. M., Raffatellu, M., Swinkels, D. W. & Tjalsma, H. Nutritional iron turned inside out: intestinal stress from a gut microbial perspective. FEMS Microbiol. Rev.38, 1202–1234 (2014).
Cherayil, B. J., Ellenbogen, S. & Shanmugam, N. N. Iron and intestinal immunity. Curr. Opin. Gastroenterol.27, 523–528 (2011).
Ganz, T. & Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol.15, 500–510 (2015).
Finlayson-Trick, E. et al. The effect of oral Iron supplementation on Gut Microbial Composition: a secondary analysis of a Double-Blind, randomized controlled trial among Cambodian women of Reproductive Age. Microbiol. Spectr.11, e05273–e05222 (2023).
Hou, K. et al. Microbiota in health and diseases. Signal. Transduct. Target. Therapy. 7, 135 (2022).
Cheng, L., Wu, H., Chen, Z., Hao, H. & Zheng, X. Gut microbiome at the crossroad of genetic variants and behavior disorders. Gut Microbes. 15, 2201156 (2023).
Wang et al. Low relative abundances of the Mucolytic Bacterium Akkermansia muciniphila and Bifidobacterium spp. in feces of children with autism. Appl. Environ. Microbiol.77, 6718–6721 (2011).
Jaeggi, T. et al. Iron fortification adversely affects the gut microbiome, increases pathogen abundance and induces intestinal inflammation in Kenyan infants. Gut. 64, 731–742 (2015).
Paganini, D., Uyoga, M. & Zimmermann, M. Iron Fortification of Foods for Infants and children in Low-Income countries: effects on the gut microbiome, gut inflammation, and Diarrhea. Nutrients. 8, 494 (2016).
Visitchanakun, P. et al. Increased susceptibility to dextran sulfate-induced mucositis of iron-overload β-thalassemia mice, another endogenous cause of septicemia in Thalassemia. Clin. Sci.135, 1467–1486 (2021).
Sae-Khow, K. et al. Pathogen-associated molecules from gut translocation enhance severity of cecal ligation and puncture sepsis in iron-overload β-thalassemia mice. J. Inflamm. Res.13, 719–735 (2020).
La Carpia, F. et al. Transfusional iron overload and intravenous iron infusions modify the mouse gut microbiota similarly to dietary iron. Npj Biofilms Microbiomes. 5, 26 (2019).
Fucharoen, S., Piankijagum, A. & Wasi, P. Deaths in beta-thalassemia/Hb E patients secondary to infections. Birth Defects Orig Artic Ser.23, 495–500 (1987).
Rahav, G. et al. Severe infections in thalassaemic patients: prevalence and predisposing factors. Br. J. Haematol.133, 667–674 (2006).
Yang, W. & Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell Mol. Immunol.18, 866–877 (2021).
Kelly, C. J. et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell. Host Microbe. https://doi.org/10.1016/J.CHOM.2015.03.005 (2015).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 341, 569–573 (2013).
Salvi, P. S. & Cowles, R. A. Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in Homeostasis and Disease. Cells. https://doi.org/10.3390/CELLS10071775 (2021).
Taher, A., Weatherall, D., Cappellini, M. & Thalassaemia Lancet doi:https://doi.org/10.1016/S0140-6736(17)31822-6. (2018).
Fernandez, M., Lokan, J., Leung, C. & Grigg, A. A critical evaluation of the role of iron overload in fatty liver disease. J. Gastroenterol. Hepatol.https://doi.org/10.1111/JGH.15971 (2022).
Hatthachote, P., Rangsin, R., Mungthin, M. & Sakboonyarat, B. Trends in the prevalence of obesity among young Thai men and associated factors: from 2009 to 2016. Military Med. Res.6, 13 (2019).
Fucharoen, S. et al. A randomized phase I/II trial of HQK-1001, an oral fetal globin gene inducer, in β-thalassaemia intermedia and HbE/β-thalassaemia. Br. J. Haematol.161, 587–593 (2013).
Rinninella, E. et al. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 7, (2019).
Morton, J. T. et al. Establishing microbial composition measurement standards with reference frames. Nat. Commun.10, 2719 (2019).
Johnson, E. L., Heaver, S. L., Walters, W. A. & Ley, R. E. Microbiome and metabolic disease: revisiting the bacterial phylum Bacteroidetes. J. Mol. Med.95, 1–8 (2017).
Fedarko, M. W. et al. Visualizing ’omic feature rankings and log-ratios using Qurro. NAR Genomics Bioinf.2, lqaa023 (2020).
Parker, B. J., Wearsch, P. A., Veloo, A. C. M. & Rodriguez-Palacios, A. The Genus Alistipes: gut Bacteria with emerging implications to inflammation, Cancer, and Mental Health. Front. Immunol.11, 906 (2020).
Gophna, U., Konikoff, T. & Nielsen, H. B. Oscillospira and related bactefrom- From metagenomic species to metabolic featmetabolismbolism of Oscillospira. Environ. Microbiol.19, 835–841 (2017).
Valles-Colomer, M. et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol.4, 623–632 (2019).
Venessa et al. <em > Butyricicoccus pullicaecorum in inflammatory bowel disease</em >. Gut. 62, 1745 (2013).
Haak, B. W. & Wiersinga, W. J. The role of the gut microbiota in sepsis. Lancet Gastroenterol. Hepatol.2, 135–143 (2017).
Jiang, H. et al. Altered gut microbiota profile in patients with generalized anxiety disorder. J. Psychiatr. Res.104, 130–136 (2018).
Kasahara, K. et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat. Microbiol.3, 1461–1471 (2018).
Sriwichaiin, S. et al. Deferiprone has less benefits on gut microbiota and metabolites in high iron-diet induced iron overload thalassemic mice than in iron overload wild-type mice: a preclinical study. Life Sci.307, 120871 (2022).
Thompson Mitchell, G., Corey Brendan, W., Yuanzheng, S. & Craft David, W. Zurawski Daniel V. Antibacterial activities of Iron Chelators against Common Nosocomial pathogens. Antimicrob. Agents Chemother.56, 5419–5421 (2012).
Reese, A. T. et al. Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. eLife. 7, e35987 (2018).
Bou-Fakhredin, R. et al. Iron overload and chelation therapy in Non-transfusion Dependent Thalassemia. Int. J. Mol. Sci. 18, 2778(2017).
Casals-Pascual, C. et al. Microbial Diversity in Clinical Microbiome studies: Sample size and statistical power considerations. Gastroenterology. 158, 1524–1528 (2020).
Li, Q. et al. Implication of the gut microbiome composition of type 2 diabetic patients from northern China. Sci. Rep.10, 5450 (2020).
Russell, S. L. et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep.13, 440–447 (2012).
Chen, Y. et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology54, 562–572 (2011).
Shan, Y., Lee, M. & Chang, E. B. The gut Microbiome and Inflammatory Bowel diseases. Annu. Rev. Med.73, 455–468 (2022).
Rizzatti, G. et al. A common factor in Human diseases. Biomed. Res. Int.2017, 1–7 (2017).
Lee, S. C. et al. Enrichment of gut-derived Fusobacterium is associated with suboptimal immune recovery in HIV-infected individuals. Sci. Rep.8, 14277 (2018).
HAMER, H. M. et al. Review article: the role of butyrate on colonic function. Aliment. Pharmacol. Ther.27, 104–119 (2008).
Wanachiwanawin, W. Infections in E-beta thalassemia. J. Pediatr. Hematol. Oncol.22, 581–587 (2000).
Zeng, M. Y., Inohara, N. & Nuñez, G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol.10, 18–26 (2017).
Zimmer, J. et al. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr.66, 53–60 (2012).
Antharam Vijay, C. et al. Intestinal dysbiosis and Depletion of Butyrogenic Bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol.51, 2884–2892 (2020).
Qin, J. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 490, 55–60 (2012).
Chen, Y. et al. High Oscillospira abundance indicates constipation and low BMI in the Guangdong gut Microbiome Project. Sci. Rep.10, 9364 (2020).
Bundgaard-Nielsen, C., Hagstrøm, S. & Sørensen, S. Interpersonal variations in gut microbiota profiles supersedes the effects of Differing Fecal Storage conditions. Sci. Rep.8, 17367 (2018).
Liang, Y. et al. Systematic analysis of impact of sampling regions and Storage methods on fecal gut Microbiome and Metabolome profiles. mSphere. 5, e00763–e00719 (2020).
Panek, M. et al. Methodology challenges in studying human gut microbiota – effects of collection, storage, DNA extraction and next generation sequencing technologies. Sci. Rep.8, 5143 (2018).
Gorzelak, M. A. et al. Methods for improving human gut Microbiome Data by reducing variability through Sample Processing and Storage of Stool. PLOS ONE. 10, e0134802 (2015).
Choo, J. M., Leong, L. E. & Rogers, G. B. Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep.5, 16350 (2015).
Cardona, S. et al. Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC Microbiol.12, 158 (2012).
Allaband, C. et al. Time of sample collection is critical for the replicability of microbiome analyses. Nat. Metabolism. 6, 1282–1293 (2024).
Jones, J., Reinke, S. N., Ali, A., Palmer, D. J. & Christophersen, C. T. Fecal sample collection methods and time of day impact microbiome composition and short chain fatty acid concentrations. Sci. Rep.11, 13964 (2021).
Johnson, A. J. et al. Daily Sampling reveals personalized Diet-Microbiome associations in humans. Cell. Host Microbe. 25, 789–802e5 (2019).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 505, 559–563 (2013).
Rinninella, E. et al. The role of diet in shaping human gut microbiota. Best Pract. Res. Clin. Gastroenterol.62–63, 101828 (2023).
Liu, Y. et al. Excess iron intake induced liver injury: the role of gut-liver axis and therapeutic potential. Biomed. Pharmacother.https://doi.org/10.1016/J.BIOPHA.2023.115728 (2023).
Koneru, S., Thiruvadi, V. & Ramesh, M. Gut microbiome and its clinical implications: exploring the key players in human health. Curr. Opin. Infect. Dis.36, 353–359 (2023).
Chancharoenthana, W., Kamolratanakul, S., Schultz, M. J. & Leelahavanichkul, A. The leaky gut and the gut microbiome in sepsis – targets in research and treatment. doi: (2023). https://doi.org/10.1042/CS20220777
Inda, M. E., Broset, E. & Lu, T. K. Fuente-Nunez, C. Emerging frontiers in Microbiome Engineering. Trends Immunol.40, 952–973 (2019). de la.
Foo, J. L., Ling, H., Lee, Y. S. & Chang, M. W. Microbiome engineering: current applications and its future. Biotechnol. J.12, 1600099 (2017).
Bai, X. et al. Engineering the gut microbiome. Nat. Reviews Bioeng.1, 665–679 (2023).
Winichagoon, P., Kumbunlue, R., Sirankapracha, P., Boonmongkol, P. & Fucharoen, S. Discrimination of various Thalassemia syndromes and iron deficiency and utilization of reticulocyte measurements in monitoring response to iron therapy. Blood Cells Mol. Dis.54, 336–341 (2015).
Schwartz, T. et al. Earth Microbiome Project (EMP) high throughput (HTP) DNA extraction protocol v1. Preprint at (2018). https://doi.org/10.17504/protocols.io.pdmdi46
Ul-Hasan, S. et al. Community ecology across bacteria, archaea and microbial eukaryotes in the sediment and seawater of coastal Puerto Nuevo, Baja California. PLOS ONE. 14, e0212355 (2019).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol.37, 852–857 (2019).
Amir et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2, 10.1128/msystems.00191 – 16 (2017).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ4, e2584 (2016).
Janssen et al. Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information. mSystems 3, (2018). https://doi.org/10.1128/msystems.00021
Bokulich Nicholas, A. et al. q2-longitudinal: Longitudinal and Paired-Sample Analyses of Microbiome Data. mSystems 3, (2018). https://doi.org/10.1128/msystems.00219
Oksanen, J. et al. vegan: Community Ecology Package. (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false Discovery rate: a practical and powerful Approach to multiple testing. J. Royal Stat. Soc. Ser. B (Methodological). 57, 289–300 (1995).
McMurdie, P. J. & Holmes, S. Phyloseq: an R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE. 8, e61217 (2013).
Funding
This work was supported by Thailand Research Fund (TRF) DPG6080003 and the Program Management Unit for Human Resources and Institution Development, Research and Innovation (B05F630062).
Author information
Authors and Affiliations
Contributions
Conceptualization: P.N., D.M, A.T., K.P., S.FMethodology: P.N., A.W., D.M, H.HH., J.C., A.T., C.N., S.T., K.P.Software: -Validation: P.N., A.W.Formal analysis: P.N., A.W., K.P.Investigation: P.N., A.W., D.M, K.P.Resources: R.K., S.FData Curation: P.N., D.M, J.C., K.P.Writing - Original Draft: P.N., A.W., H.HH., K.P.Writing - Review & Editing: P.N., S.FVisualization: P.N., A.W., H.HH.Supervision: D.M, A.T., C.N., S.T., R.K., S.FProject administration: P.N., K.P., S.F Funding acquisition: S.F.
Corresponding author
Ethics declarations
Competing interests
All authors have read and agreed to the published version of the manuscript. D.M. is a consultant for, and has equity in, BiomeSense, Inc. The terms of these arrangements have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies. Rob Knight is a scientific advisory board member, and consultant for BiomeSense, Inc., has equity and receives income. He is a scientific advisory board member and has equity in GenCirq. He is a consultant for DayTwo, and receives income. He has equity in and acts as a consultant for Cybele. He is a co-founder of Biota, Inc., and has equity. He is a cofounder of Micronoma, and has equity and is a scientific advisory board member. The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Ethical statement
The study was approved by the Mahidol University Central Institutional Review Board (MU-CIRB) (Approved protocol number MU-CIRB 2018/181.1309 and COA No. MU-CIRB 2018/184.2610). MU-CIRB is in full compliance with International Guidelines for Human Research Protection including Declaration of Helsinki, The Belmont Report, CIOMS Guidelines and the International Conference on Harmonization in Good Clinical Practice (ICH-GCP). Informed consent was obtained from all subjects involved in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nonejuie, P., Wilantho, A., McDonald, D. et al. Differential gut microbiota composition in β-Thalassemia patients and its correlation with iron overload. Sci Rep 14, 23858 (2024). https://doi.org/10.1038/s41598-024-75456-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-75456-4
Keywords
This article is cited by
-
Gut bacterial dysbiosis in pediatric severe malaria associates with post-discharge mortality
Nature Communications (2025)
-
Gut microbiota-derived TMAO and SIRT1/HMGB1 Axis: unveiling mechanisms of renal impairment in beta-thalassemia major
Pediatric Research (2025)
-
Phage therapy and the microbiome in hematologic malignancies: opportunities, mechanisms, and early evidence
Journal of Cancer Research and Clinical Oncology (2025)
