
Abstract
Ruminants have the ability to digest human-inedible plant materials, due to the symbiotic relationship with the rumen microbiota. Rumen microbes supply short chain fatty acids, amino acids, and vitamins to dairy cows that are used for maintenance, growth, and lactation functions. The main goal of this study was to investigate gene-microbiome networks underlying feed efficiency traits by integrating genotypic, microbial, and phenotypic data from lactating dairy cows. Data consisted of dry matter intake (DMI), net energy secreted in milk, and residual feed intake (RFI) records, SNP genotype, and 16S rRNA rumen microbial abundances from 448 mid-lactation Holstein cows. We first assessed marginal associations between genotypes and phenotypic and microbial traits through genomic scans, and then, in regions with multiple significant hits, we assessed gene-microbiome-phenotype networks using causal structural learning algorithms. We found significant regions co-localizing the rumen microbiome and feed efficiency traits. Interestingly, we found three types of network relationships: (1) the cow genome directly affects both rumen microbial abundances and feed efficiency traits; (2) the cow genome (Chr3: 116.5 Mb) indirectly affects RFI, mediated by the abundance of Syntrophococcus, Prevotella, and an unknown genus of Class Bacilli; and (3) the cow genome (Chr7: 52.8 Mb and Chr11: 6.1–6.2 Mb) affects the abundance of Rikenellaceae RC9 gut group mediated by DMI. Our findings shed light on how the host genome acts directly and indirectly on the rumen microbiome and feed efficiency traits and the potential benefits of the inclusion of specific microbes in selection indexes or as correlated traits in breeding programs. Overall, the multistep approach described here, combining whole-genome scans and causal network reconstruction, allows us to reveal the relationship between genome and microbiome underlying dairy cow feed efficiency.
Similar content being viewed by others

Introduction
Like all ruminants, dairy cows have a symbiotic relationship with their rumen microbiota. The cow provides a favorable environment (i.e., the rumen) for microbial growth and survival. In turn, the rumen microbes digest and transform feedstuffs into volatile fatty acids and synthesize amino acids and vitamins that are absorbed to support maintenance, growth, and lactation1. The effectiveness of the process to convert feed into milk and body tissue is what we know as feed efficiency, which greatly impacts the profitability and sustainability of the dairy industry. Feed efficiency is commonly measured using residual feed intake (RFI), defined as the difference between the actual dry matter intake (DMI) and predictions from regression on known energy sinks, including metabolic body weight, change in body weight, and milk energy2,3. There are multiple benefits to improving dairy cow feed efficiency, including reducing the environmental impact of dairy farming and improving dairy farm profitability4.
The cow genome exerts moderate control over feed efficiency and low to moderate control over rumen microbial abundances. Indeed, feed efficiency traits in Holstein cows show heritability estimates that range from 0.16 for RFI to 0.20 for DMI5. Similarly, rumen microbial abundances have heritability estimates that range from 0.10 to 0.406,7, and these differences in microbial populations also contribute to feed efficiency at the whole animal level8. It was recently shown that the rumen microbiome could change the total heritability (i.e., direct and indirect additive genetic effects) of feed efficiency traits through a mediated genetic effect. The change in total heritability depends on the phenotypic effect of microbes on feed efficiency and the genetic correlations between microbes and observed traits7. The major causes of genetic correlations due to direct host effects on traits are the linkage disequilibrium between close genomic regions and the presence of pleiotropic genes in the host genome that influence more than one trait9.
Whole-genomic scans have successfully identified genomic regions associated with many traits in US Holstein cows10. Genomic scans for feed efficiency traits, such as RFI and DMI, showed no large peaks or significant quantitative trait loci (QTL)11,12, suggesting polygenic inheritance. Similarly, recent genomic scans for the rumen microbiome have not detected large QTL effects, but these studies have struggled with dimensionality (numerous microbes with various potential QTL for evaluation). Note that the co-localization of rumen microbial QTL (rmQTL) with phenotypic QTL (pQTL) from marginal associations between genotypes and microbial and phenotypic traits through genomic scans, respectively, could help to understand the relationship between the host genome, microbiome, and phenotypes. It seems reasonable to view this complex relationship as a system or network. In this context, Bayesian networks are a convenient way to represent complex patterns of relationships13. They provide a high-level, holistic view of the interplay between random variables (e.g., genotypes, microbes, and traits) by representing them as nodes and linking them with edges that describe their interactions. Hence, the integration of phenotypic data with the host genome and rumen microbiome information via causal networks has the potential to shed light on the genome-by-microbiome interactions that underly dairy cow feed efficiency.
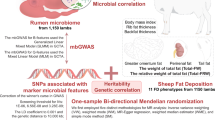
This study aimed to reveal gene-microbiome networks underlying feed efficiency integrating genotypic, microbial, and phenotypic data obtained from mid-lactation dairy cows. Similar to the approach presented by Peñagaricano et al.14, we first explored marginal associations between genotypes and microbial and phenotypic traits through genomic scans. Then, in regions where several rmQTL and pQTL co-localized, we performed network reconstruction using causal structural learning algorithms.
Materials and methods
Ethics statement
Animal trials were conducted at the University of Florida (Gainesville, FL, USA) and the University of Guelph (Guelph, ON, Canada) in accordance with relevant guidelines and regulations. The Institutional Animal Care Committees of Florida (protocol no 202300000187) and Guelph (protocol no 4064) approved the experimental protocols. No animal procedures in this study required the use of anesthesia or euthanasia, as all animal handling and sampling methods compiled with animal welfare standards. Reporting in this manuscript follows the recommendations of the ARRIVE guidelines (https://arriveguidelines.org).
Animals
We used a total of 451 lactating Holstein cows from two research farms. The cows were enrolled in six experiments: 237 cows from five experiments conducted at the University of Florida Dairy Research Unit (Alachua, FL, USA) and 214 cows from a single experiment at the Ontario Dairy Research Centre (Elora, ON, Canada), between January 2019 and July 2020. Overall, this study included 221 primiparous and 230 multiparous cows, ranging from 31 to 136 days in milk (DIM), with an average body weight of 657 \(\:\pm\:\) 82 kg.
In Florida, cows were housed in sand-bedded free-stall barns and were randomly assigned to individual Calan gates (American Calan Inc., Northwood, NH, USA) to evaluate daily feed intake. Cows had ad libitum access to diets that were fed as total mixed rations (TMR). The TMR were weighed and fed twice daily approximately at 06: 00 and 13: 00 and refusals were weighed once daily, before morning feeding. The amount of TMR offered to individual cows was adjusted daily to result in 5 to 10% refusals. Cows were weighed twice daily, immediately after each milking, on a walk-over scale (AfiWeigh, SAE Afikim, Israel) as they left the milking parlor. In Ontario, cows were housed in a free-stall barn equipped with Roughage Intake Control System (Hokofarm Group, Marknesse, The Netherlands) and mattress-beds with chopped wheat straw bedding. The TMR was delivered twice daily approximately at 09: 00 and 15: 30. Refusals were removed from the feed bins daily, and the amount of feed offered was adjusted daily to allow 8% refusals. Cows were weighed twice daily on a walk-over scale (DeLaval, Tumba, Sweden).
Cows were milked twice daily, and the milk yield was recorded using electronic milk flow meters (AfiFlo, SAE Afikim, USA; and DeLaval, Canada). Milk samples were collected once (in Canada) or twice (in Florida) weekly from both milkings for fat, protein, and lactose analyses, performed at the DHIA testing laboratory (Lactanet, Guelph, ON, Canada) and at the Southeast Milk Inc. DHI Laboratory (Belleview, FL, USA). For Canada, the lactose percentage was calculated based on the tank average for each week.
Phenotypic data
We evaluated three feed efficiency traits: DMI, net energy secreted in milk (NESec), and RFI. Net energy secreted in milk (Mcal/day) was calculated according to the NRC equations for nutrient requirements of dairy cattle15 as follows:
We generated weekly means of body weight (BW) using daily records. Weekly changes in BW were calculated as the difference between two consecutive weeks and divided by the number of days between measurements to generate a daily change in BW in kg/day.
The DMI records were regressed on the three main energy sinks to calculate RFI values using a linear model as follows:
where \(\:\varvec{m}\varvec{D}\varvec{I}\varvec{M}\) represents the fixed effect of midpoint DIM for the period from 31 to 136 DIM (2 levels: ≤ 67 DIM and > 67 DIM), \(\:\varvec{c}\varvec{o}\varvec{h}\varvec{o}\varvec{r}\varvec{t}\) represents the fixed effect of treatment nested within experiment and season (13 levels: 5 levels for Florida and 8 levels for Canada), \(\:\varvec{p}\varvec{a}\varvec{r}\varvec{i}\varvec{t}\varvec{y}\) represents the fixed effect of lactation number with four levels (1, 2, 3, and 4+), \(\:NESec\) is the net energy secreted in milk with partial fixed regression coefficient \(\:{\varvec{\beta\:}}_{1},\) \(\:mBW\) is the metabolic BW with partial fixed regression coefficient \(\:{\varvec{\beta\:}}_{2}\), \(\:\varDelta\:BW\) is the change in BW with partial fixed regression coefficient \(\:{\varvec{\beta\:}}_{3}\), and \(\:e\) is the random residual. The residual of this model is defined as RFI, for which positive values indicate animals that had DMI above what was expected, i.e., less feed efficient animals, whereas negative values indicate animals with DMI below expected, i.e., more feed efficient animals. The coefficient of determination of the model was 0.81. All variables included in the model significantly (P-value < 0.05) contributed to variation in DMI.
Genotypic data
Cows were genotyped with either low (20k) or medium (50k) density single nucleotide polymorphisms (SNP) chips (Illumina Inc., San Diego, CA, USA) and then imputed to a total of 78,933 SNP across the cow genome. The genotype data were kindly provided by the Council on Dairy Cattle Breeding (Bowie, MD, USA). We retained 78,016 SNP after quality control, including call rates of ≥ 95% for SNP and ≥ 95% for individuals, and excluding SNP with minor allele frequency < 1% and in the sexual chromosome. Marker information, including chromosome and position, was obtained according to the Bos taurus reference genome assembly ARS-UCD1.3.
Microbial data
Rumen fluid sampling was performed on a single day in the middle of each experimental period when cows were between 66 and 105 DIM. The rumen fluid sample for each cow was obtained using an orogastric tube attached to a glass vial and equipped with a pump. Samples were snap-frozen in liquid nitrogen immediately after sample collection. The DNA extraction and library preparation were processed through a 96-channel portable robot pipette to improve pipetting precision. On the day of DNA extraction, rumen content samples were thawed in ice, and DNA was extracted using a Mag-Bind® Universal pathogen 96 Kit (Omega Bio-Tek Norcross, GA, USA) according to manufacturer instructions. The 16S rRNA gene V4 region was amplified by PCR amplification using the following primers: forward (515F-Y, 5’-GTGYCAGCMGCCGCGGTAA) and reverse (806R, 5’-GGACTACNVGGGTWTCTAAT) as described by Caporaso et al.16 and modified by Apprill et al.17 and Parada et al.18. After DNA amplification, the presence and size of amplicons were verified through gel electrophoresis using a 1.2% (wt/vol) agarose gel stained with 0.5 mg/mL ethidium bromide. Purification of amplified DNA was performed through magnetic Mag-Bind® TotalPure Next Generation Sequencing (Omega Bio-Tek Norcross, GA, USA) following manufacturer instructions. The DNA concentration and purity was assessed through spectrophotometry, samples were then diluted to the same concentration using ultrapure distilled water, and equal volumes of individual samples were pooled together for sequencing. A final accurate and precise double-stranded DNA quantification was performed in a Qubit® fluorometric machine. For sequencing, the pooled library was diluted to 4 nM, denatured, and combined with a PhiX Control 3 following MiSeq System Denature and Dilute Libraries Guide (Illumina, San Diego, CA, USA). A MiSeq Reagent Kit v2 of 300 cycles (Illumina, San Diego, CA, USA) was used in an Illumina MiSeq platform set for a 16 S Metagenomics Workflow. The raw sequence reads were deposited in the Sequence Read Archive of the National Center for Biotechnology Information under the Bio Project PRJNA962991.
Amplicon sequences of the 451 rumen samples were processed using the DADA2 pipeline version 1.819 in the R software20. Briefly, denoising analysis was performed by demultiplexing sequencing reads and inspecting for errors. Sequences were trimmed and filtered, chimeras were removed, and an amplicon sequence variant (ASV) table with 9707 ASV was created. Taxonomy assignment was performed using 16 S rRNA SILVA version 138 database up to the genus level21. We discarded one sample during wet lab processing and two samples with very few (< 3361) sequences and few (< 120) ASV. We retained 448 samples and 1,636 ASV after the removal of those ASV present in less than 5% of samples. Considering the compositionality of the microbial abundances, 1 was added to all counts in the abundance table to avoid zeros and then applied a centered log-ratio transformation (CLR) with the function clr implemented in the R package compositions22.
Candidate rumen microbes
Rumen microbiome data have many variables, i.e., 1636 ASV, so we pre-selected rumen microbes that can potentially be used in selective breeding, as shown in Martinez-Boggio et al.7. Using recursive models to evaluate the mediation effect of the rumen microbiome on feed efficiency, we ended up selecting 272 microbes with moderate heritability (\(\:{h}^{2}>0.10\)), significant phenotypic effects on DMI, NESec, and/or RFI, and genetically correlated with DMI, NESec, and/or RFI (see Supplementary Table S1 online).
Genomic scans
Two complementary genome scans were performed: first, we carried out a classical phenotypic QTL scan integrating phenotypic and genotypic data, and second, we performed a rumen microbial QTL scan integrating the abundances of the 272 relevant microbes and genotypic data. For the pQTL and rmQTL scans, the following linear model was fitted separately to each phenotype and microbial trait:
where \(\:\varvec{y}\) is the phenotypic trait (DMI, NESec, and RFI) or microbial trait (1 to 272 ASV abundances), \(\:\varvec{c}\varvec{o}\varvec{h}\varvec{o}\varvec{r}\varvec{t}\) represents the fixed effect of the cohort, as treatment nested within experiment and season (13 levels), \(\:\varvec{p}\varvec{a}\varvec{r}\varvec{i}\varvec{t}\varvec{y}\) represents the fixed effect of the lactation number with four levels (1, 2, 3, and 4+), \(\:SNP\) is the additive SNP effect (coded as 0, 1, or 2, i.e., the number of reference allele in the SNP genotype) with partial fixed regression coefficient \(\:{\varvec{\beta\:}}_{\varvec{S}\varvec{N}\varvec{P}}\), \(\:\varvec{c}\varvec{o}\varvec{w}\) is the vector of additive polygenic effects, and \(\:\varvec{\epsilon\:}\) is a vector of residual effects. The assumption of the model is \(\user2{cow}\sim N\left( {0,\user2{G}\sigma _{g}^{2} } \right)\), where \(\:\varvec{G}\) is the additive genomic relationship matrix, computed as \(\:\varvec{G}=\varvec{Z}{\varvec{Z}}^{\varvec{{\prime\:}}}/k\), where \(\:\varvec{Z}\) is a matrix of centered and standardized SNP genotypes and \(\:k\) represents the number of SNP23. The WOMBAT software24 was used for model fitting and estimating SNP solutions. To correct for multiple testing, we applied the simpleM method25. This approach consisted of four steps: (1) for each chromosome, calculate a correlation matrix for the SNP using the composite linkage disequilibrium (LD); (2) compute the eigenvalues through principal component analysis of the composite LD matrix; (3) calculate the effective number of SNP per chromosome as the number of principal components required to explain 99% of the variance in the SNP. Because SNP on different chromosomes are expected to be in linkage equilibrium in the general population, the total effective number of SNP (Meff) is obtained by summing the effective numbers of SNP of all chromosomes. And finally, (4) adjust P-values from single SNP association tests for multiple comparisons using Šidák correction based on Meff: adjusted P-value \(\:=1-{\left(1-Pvalue\right)}^{Meff}.\) Genes located near significant QTL were retrieved from the Ensembl database26 using the BioMart web interface, which is based on the Bos taurus genome assembly ARS-UCD1.3.
Causal structural learning
We reconstructed causal networks between cow genome, rumen microbiome, and feed efficiency combining genotypes (i.e., co-localized SNP between microbiome and feed efficiency), microbial abundances, and phenotypes (DMI, NESec, or RFI).
We represented the causal structures using graphical models because they combine the rigor of a probabilistic approach with the intuitive representation of relationships given by graphs. Graphical models are composed of two parts: a set \(\:\varvec{V}=({v}_{1},{v}_{2},\dots\:,{v}_{k})\) of random variables describing the quantities of interest, and a graph \(\:G=(\varvec{V},\varvec{E})\), where \(\:\varvec{V}\) and \(\:\varvec{E}\) represent sets of vertices and edges, respectively. Each vertex (or node) in \(\:G\) represents one of the variables in the network, and the set of edges (or links) connecting variables is used to express the dependence structure of the data13. We used the Incremental Association Markov Blanket (IAMB) algorithm27 for the network structure learning or reconstruction. The IAMB algorithm first learns the Markov Blanket of each variable in the dataset; the Markov Blanket of a given variable \(\:{v}_{i}\) is defined as the minimal set of variables conditioned on which all other variables are probabilistically independent of the target variable \(\:{v}_{i}\). This preliminary step reduces the number and the size of the subsets considered in the conditional tests of network reconstruction, and hence results in a lower computational complexity without compromising the accuracy of the resulting inferred network27. In the context of normally distributed variables, these tests are functions of the partial correlation coefficients between each pair of variables in \(\:\varvec{V}\), conditional on a subset of variables \(\:\varvec{W}\in\:\varvec{V}\), represented by \(\:{p}_{i{i}^{{\prime\:}}|\varvec{W}}\). Here, we used the Fisher’s Z test, which involves a transformation of the linear correlation coefficient and is defined as:
where \(\:{\widehat{\rho\:}}_{i{i}^{{\prime\:}}|\varvec{W}}\) is the estimated of the partial correlation, and \(\:\left|\varvec{W}\right|\) denotes the number of variables in set \(\:\varvec{W}\). The \(\:Z\) statistics has an approximate normal distribution with mean zero and variance 1, i.e., \(Z\left( {v_{i} ,v_{{i^{\prime}}} {\text{|}}\user2{W}} \right)\sim N\left( {0,1} \right)\). After the structure of the network was learned, the estimation of the parameters of the local distributions was performed using maximum likelihood, where the causal parameters take the form of regression coefficients. Furthermore, the stability of the structure of the causal networks was evaluated using Jackknife resampling. By leaving out one observation per time from the dataset, we could evaluate the stability of each edge in the original network in terms of presence (presence or absence in the resampled network) and direction (same direction as the original edge, opposite direction, or undirected arc). All these network analyses were performed using the R package bnlearn28.
Results
Genomic scans
We performed classical whole-genome scans integrating phenotypes, namely DMI, NESec, and RFI, with genotypic information (78,016 SNP). We identified three significant pQTL (adjusted P-value \(\:\le\:\) 0.10) for DMI, one for NESec, and two for RFI, as shown in Fig. 1. The strongest SNP effects for DMI were found in regions 6.1–6.2 Mb on BTA11 and 7.4 Mb on BTA23, and for RFI were clustered in regions 116.5-117.3 Mb on BTA3 and 28.1–30.5 Mb on BTA16. Note that for NESec we detected many suggestive SNP (adjusted P-value \(\:\le\:\) 0.22) on five chromosomes (see Supplementary Table S2 online).

Genomic scans for dry matter intake (DMI), net energy secreted in milk (NESec), and residual feed intake (RFI). The horizontal line indicates the adjusted P-value threshold of 0.10.
We also performed genomic scans integrating rumen microbial abundances with genotypic information (78,016 SNP). We obtained 1,028 significant rmQTL (adjusted P-value \(\:\le\:\) 0.10) mapped all across the cattle genome but mainly on BTA5 (66 rmQTL), BTA11 (49 rmQTL), BTA9 (49 rmQTL), and BTA7 (49 rmQTL) (see Supplementary Table S2 online). The rumen microbial genera with more significant QTL were Prevotella (164 rmQTL), Prevotella_7 (71 rmQTL), Rikenellaceae RC9 gut group (46 rmQTL), Acetitomaculum (42 rmQTL), and Christensenellaceae R-7 group (36 rmQTL). Note that the Prevotella genus, represented by more than 50 ASV, showed associations all across the genome. However, for some microbial genera, we identified strong SNP effects on specific genomic regions (Fig. 2). For Prevotella_7 we found five ASV with significant SNP on three regions of BTA11 (1.0-1.3, 2.6–2.9, and 6.1–6.2 Mb), and for Acetitomaculum we found four ASV with significant SNP on two regions of BTA5 (61.8–65.6 and 106.2-107.2 Mb). Also, for Prevotellaceae UCG-004 (ASV1018) we found SNP effects only on BTA5 and BTA11. Furthermore, we detected 69 significant co-localized SNP on 23 chromosomes for rumen microbiome. We identified one SNP on BTA29 co-localizing for 13 ASV belonging to the genera Prevotella and Prevotella_7, and the Lachnospiraceae family. Also, we identified four SNP co-localized in the region 1.0-1.3 and 6.1–6.2 Mb on BTA11 for six ASV from Prevotella_7, Prevotellaceae UCG-004, Lachnospiraceae NK3A20 group, and [Eubacterium] nodatum group.

Genomic scans for rumen microbial abundances. The horizontal line indicates the adjusted P-value threshold of 0.10.
Remarkably, we obtained two significant co-localized SNP between DMI and rumen microbiome clustered in the region 6.1–6.2 Mb on BTA11 (Fig. 3). Note that we also found very close significant SNP effects between feed efficiency traits and rumen microbial traits in eight chromosomes (Table 1). For RFI, we detected three pQTL: one in the region 116.5-117.3 Mb on BTA3, where Prevotella ASV57, Syntrophococcus ASV666, and [Eubacterium] brachy group ASV2236 showed significant SNP; one in the region 28.1–30.5 Mb on BTA16, where Succiniclasticum ASV83 showed a significant SNP; and one in the region 37.2 Mb on BTA19, where Acetitomaculum ASV1936 showed a significant SNP. Furthermore, for trait NESec, we found significant SNP effects for F082 ASV537 and Lachnobacterium ASV1454 close to the suggestive pQTL in the region 16.5 Mb on BTA13.

Co-localized quantitative trait loci for phenotypes (dry matter intake (DMI) and residual feed intake (RFI)) and rumen microbial abundances. The horizontal line indicates the adjusted P-value threshold of 0.10.
Causal networks
The pQTL and rmQTL analyses showed that some feed efficiency traits and rumen microbes are associated with the same SNP on BTA11. Also, there are co-localized regions on BTA3 and BTA7 with significant pQTL and rmQTL. So, to infer the functional relationship involving the cow genome, rumen microbiome, and feed efficiency traits, we used the IAMB algorithm in conjunction with Fisher’s \(\:Z\) test to assess for conditional independence (\(\:\alpha\:=0.10\)). The causal structural learning was performed using adjusted phenotypic and microbial traits (i.e., corrected by cohort and parity effects), and the most significant SNP located on BTA3, BTA7, and BTA11. We used as prior knowledge that the genotypes may have a causal effect on rumen microbiome and feed efficiency but not the opposite or between genotypes. The stability of the network was evaluated using Jackknife resampling and expressed as the frequency at which a given edge was presented or not and with the same direction in the resampled network. Figure 4 shows that the stability and direction of the edges in the causal networks were very consistent, i.e., most of the edges were present in more than 65% of the resampled networks.

Casual networks integrating genotypic data (blue), rumen microbial abundances (green), and phenotypic traits (orange). (a) causal network showing a direct and indirect relationship between genome and residual feed intake (RFI) mediated by the rumen microbiome; (b) causal network showing a direct relationship between genome, phenotypes (dry matter intake (DMI), and net energy secreted in milk (NESec)), and rumen microbiome, and indirect with the rumen microbiome and NESec mediated by DMI; (c) causal network showing a direct relationship between genome, DMI, and rumen microbiome, and indirect with the rumen microbiome mediated by DMI. The consistency of the network structure is expressed in terms of stability (the frequency at which a given edge is presented) and direction (the frequency at which a given edge has the same direction; in parenthesis) in the resampled networks comparing with the original network.
Interestingly, we identified different types of causal relationships between variables, with direct and indirect pathways. Figure 4 shows that two QTL on BTA3 (labeled as Chr3:116,575,934 and Chr3:117,335,980) were directly and indirectly associated with RFI mediated by the rumen microbiome (ASV1047, ASV666, and ASV57). Furthermore, two QTL on BTA7 (Chr7:17,454,617 and Chr7:52,843,633) showed direct effects on DMI and NESec, and an indirect effect on Rikenellaceae RC9 gut group (ASV460) and CAG-352 (ASV1085) mediated by DMI. We also found mediated effects of the cow genome on the rumen microbiome by DMI on BTA11. The two co-localized SNP (labeled as Chr11:6,192,173 and Chr11:6,216,461) between DMI and rumen microbiome were marginally associated with DMI and six rumen microbes. Note that DMI is mediating genetic effects on Rikenellaceae RC9 gut group (ASV31). We also found in this network marginal associations between microbial genera such as Prevotella_7, Rikenellaceae RC9 gut group, and the Lachnospiraceae family.
Discussion
We explored the relationships between the host genome and rumen microbiome underlying the expression of feed efficiency traits in mid-lactation Holstein cows. Cows establish a symbiotic relationship with the rumen microbiome, which allows them to digest human-inedible plant materials, supplying 70% of the energy the host needs and 60% of the amino acids that reach the small intestine29. Given this critical role, the rumen microbiome is widely recognized for its major contributions to feed efficiency7,30,31. Recently, we evaluated the mediation effects of rumen microbial abundances on feed efficiency traits using recursive models7. However, how the cow genome, rumen microbiome, and feed efficiency traits (i.e., DMI, NESec, and RFI) interact remain unclear.
Feed efficiency traits showed no large peaks or pQTL in our genomic scans, which suggests they are highly polygenic traits regulated by many small-sized effects. These findings agree with previous studies in Holstein cows in which authors did not find specific genomic regions associated with RFI11,12. Residual feed intake is a composite trait including major energy sinks (e.g., energy needs for milk synthesis and cow’s maintenance), so it is related to diverse biological processes and metabolic pathways12 that could result in a diluted effect of many associated genomic regions. Contrary to RFI, we expected to find genomic regions associated with NESec (calculated using fat and true protein), mainly on BTA14, due to the presence of a major gene named DGAT132. Note that these results could be explained by the limited sample size of 448 cows used in our study. Also, for DMI, we did not find clear pQTL in genomic regions where previous studies had detected significant effects for DMI11,12 and fertility and economic merit10,33.
Rumen microbial abundances showed many rmQTL across the genome, as reported in different livestock species34,35,36,37. These findings suggest that many small-sized gene effects also regulate the rumen microbiome. However, we identified significant host genetic effects on the abundance of the genera Acetitomaculum, Prevotella_7, and Prevotellaceae UCG-004 on BTA5 and BTA11, Treponema on BTA8, and Butyrivibrio on BTA7 (Fig. 2). Note that many of these microbial genera are generalists, possessing broad functional capability that allow them to adapt to diverse ecological niches38. Tapio et al.39 reported the presence of Acetitomaculum, Butyrivibrio, and Prevotella within multiple rumen microbial sub-networks in dairy cows. Interestingly, these microbes co-localize, which means that the same host genomic regions regulate their abundance. Hence, we hypothesize that major host genomic regions control the abundance of these generalist microbes through physical and metabolic mechanisms, including passage rate, constant supply of substrates, pH, temperature, among others40. We did not find any major gene that could directly explain the mechanisms the host genome has to control microbial abundances in the rumen.
Furthermore, we found co-localization of feed efficiency traits and rumen microbial abundances on many chromosomes. Note that all the rumen microbes included in the genomic scans were heritable and correlated with DMI, NESec, and/or RFI (see Supplementary Table S1 online). The identification of genomic regions shared by both microbiome and feed efficiency traits, along with our previous findings that the inclusion of specific microbes into genomic models can increase the total heritability of feed efficiency traits7, highlights the potential use of microbiome information in selective breeding. Rumen microbes could be used as correlated traits or even included in selection indexes.
We further explored these genomic regions using Bayesian networks to decipher potential causal relationships between the host genome and rumen microbiome that could be underlying feed efficiency traits. Remarkably, the structural learning algorithm detected different types of relationships, as shown in Fig. 4. First, the cow genome directly affects feed efficiency traits and rumen microbiome abundances, which agrees with the genomic scans results. Second, the cow genome (Chr3:116,575,934) indirectly affects RFI, mediated by the abundance of Syntrophococcus, Prevotella, and an unknown genus of Class Bacilli. Prevotella genus plays a major role in the rumen due to its diverse metabolic roles, such as its ability to utilize starch, protein, hemicellulose, and pectin to produce acetate, succinate, and propionate39. Our findings show that Prevotella interacts with other microorganisms and impacts dairy cow feed efficiency. Third, the cow genome on BTA7 (Chr7:52,843,633) and BTA11 (Chr11:6,192,173 and Chr11:6,216,461) affects the abundance of Rikenellaceae RC9 gut group mediated by DMI and the Lachnospiraceae family (ASV245). Rikenellaceae RC9 gut group produce propionate, acetate, and/or succinate, and it was observed that the diet affected its associations with growth performance and feed efficiency39,41. Interestingly, this network shows that the direction typically assumes in the recursive model (\(\:ASV\to\:DMI\))7 could be in the opposite direction (\(\:DMI\to\:ASV\)). These findings highlight potential mechanisms through which the host can control the rumen microbiome, such as passage rate. Note that less feed intake will increase the retention time in the rumen, i.e., feed stays longer, which in turn can increase digestibility per unit of organic matter consumed, and that could affect rumen turnover, the rate of microbial growth, and, therefore, affect the rumen microbial abundances and RFI. Notably, research has shown that lactating cows that eat at a slower rate are more feed efficient5. Moreover, our results also show the complexity of the microbial interactions in the rumen involving microbes from different families (Prevotellaceae, Rikenellaceae, and Lachnospiraceae) interacting with each other due to their broad metabolic capabilities.
We revealed host genome and rumen microbiome networks underlying feed efficiency traits in Holstein cows. These findings, derived from relevant rumen microbes with a moderate heritability, significant phenotypic effect on feed efficiency, and genetic correlation with feed efficiency traits, add significant biological evidence to support their use in breeding programs. We acknowledge that the use of a reduced number of candidate microbes might be too simplistic and may not fully account for the dimensionality of the rumen microbiome. However, to our knowledge, this study is unique because it employs Bayesian networks to study the direct and indirect relationships between cow genome, rumen microbiome, and complex phenotypic traits in dairy cattle.
Structural equation models and graphical models have been used to infer causal relationships between variables42,43,44,45 and genomic scans are widely used to reveal the genetic basis of complex human46 and animal47 traits. Here, we described a multistep procedure for inferring causal networks integrating multi-omics data, including microbiome data. Such approach can be powerful for revealing complex gene-microbiome interactions underlying complex phenotypic traits in livestock species.
Overall, our findings shed light on how the cow genome can act directly on both the rumen microbiome and feed efficiency, and indirectly through DMI on Rikenellaceae RC9 gut group, and through the rumen microbiome on RFI. Furthermore, the multistep procedure that combines genome scans for phenotypes and microbiome and Bayesian networks to integrate the multi-omics data allowed us to reveal different types (direct and indirect paths) of relationships between the cow genome and rumen microbiome underlying feed efficiency.
Data availability
The raw sequence reads were deposited in the Sequence Read Archive of the National Center for Biotechnology Information under the Bio Project PRJNA962991.
References
Cammack, K. M., Austin, K. J., Lamberson, W. R., Conant, G. C. & Cunningham, H. C. Ruminnat nutrition symposium: tiny but mighty: The role of the rumen microbes in livestock production. J. Anim. Sci. 96, 752–770 (2018).
Berry, D. P. & Crowley, J. J. Cell biology symposium: Genetics of feed efficiency in dairy and beef cattle. J . Anim. Sci. 91, 1594–1613 (2013).
VandeHaar, M. J. et al. Harnessing the genetics of the modern dairy cow to continue improvements in feed efficiency. J. Dairy. Sci. 99, 4941–4954 (2016).
Pryce, J. E., Wales, W. J., De Haas, Y., Veerkamp, R. F. & Hayes, B. J. Genomic selection for feed efficiency in dairy cattle. Animal. 8, 1–10 (2014).
Cavani, L. et al. Estimates of genetic parameters for feeding behavior traits and their associations with feed efficiency in Holstein cows. J. Dairy. Sci. 105, 7564–7574 (2022).
Difford, G. F. et al. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PLoS Genet. 14, e1007580 (2018).
Martinez-Boggio, G. et al. Investigating relationships between the host genome, rumen microbiome, and dairy cow feed efficiency using mediation analysis with structural equation modeling. J. Dairy. Sci. 107, 8193–8204 (2024).
Martinez Boggio, G. et al. Host and rumen microbiome contributions to feed efficiency traits in Holstein cows. J. Dairy. Sci. 107, 3090–3103 (2024).
Bourdon, R. Chapter 13 - Correlated response to selection. In understanding animal breeding (ed Linsner, K.) 275–276 (Prentice-Hall, Upper Saddle River, New Jersey, (2000).
Cole, J. B. et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genom. 12, 408 (2011).
Lu, Y. et al. Genome-wide association analyses based on a multiple-trait approach for modeling feed efficiency. J. Dairy. Sci. 101, 3140–3154 (2018).
Li, B. et al. High-density genome-wide association study for residual feed intake in Holstein dairy cattle. J. Dairy. Sci. 102, 11067–11080 (2019).
Scutari, M. & Denis, J. B. Bayesian Netw. (Chapman and Hall/CRC, 2021).
Peñagaricano, F. et al. Exploring causal networks underlying fat deposition and muscularity in pigs through the integration of phenotypic, genotypic and transcriptomic data. BMC Syst. Biol. 9, 58 (2015).
National research council. Nutr. Requirements Dairy Cattle (National Academies, 2001).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012).
Apprill, A., Mcnally, S., Parsons, R. & Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137 (2015).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 13, 581–583 (2016).
R Core Team. A language and environment for statistical computing. Preprint at. (2022).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D591–D596 (2013).
van den Boogaart, K. G. & Tolosana-Delgado, R. Compositions’: A unified R package to analyze compositional data. Comput. Geosci. 34, 320–338 (2008).
VanRaden, P. M. Efficient methods to compute genomic predictions. J. Dairy. Sci. 91, 4414–4423 (2008).
Meyer, K. WOMBAT: A tool for mixed model analyses in quantitative genetics by restricted maximum likelihood (REML). J. Zhejiang Univ. Sci. B. 8, 815–821 (2007).
Gao, X., Starmer, J. & Martin, E. R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 32, 361–369 (2008).
Martin, F. J. et al. Ensembl 2023. Nucleic Acids Res. 51, D933–D941 (2023).
Tsamardinos, I., Aliferis, C. & Statnikov, A. Algorithms for large scale markov blanket discovery. In: Proc. of the 16th International Florida Artificial Inteligence Research Society Conference AAAI Press, Nenlo Park, California, (2003).
Scutari, M. Learning Bayesian networks with the bnlearn R package. (2009).
Seymour, W. M., Campbell, D. R. & Johnson, Z. B. Relationships between rumen volatile fatty acid concentrations and milk production in dairy cows: A literature study. Anim. Feed Sci. Technol. 119, 155–169 (2005).
Monteiro, H. F. et al. Rumen and lower gut microbiomes relationship with feed efficiency and production traits throughout the lactation of Holstein dairy cows. Sci. Rep. 12, 4904 (2022).
Delgado, B. et al. Whole rumen metagenome sequencing allows classifying and predicting feed efficiency and intake levels in cattle. Sci. Rep. 9, 11 (2019).
Grisart, B. et al. Genetic and functional confirmation of the causality of the DGAT1 K232A quantitative trait nucleotide in affecting milk yield and composition. Proc. Natl. Acad. Sci. 101, 2398–2403 (2004).
Cole, J. B. et al. Distribution and location of genetic effects for dairy traits. J. Dairy. Sci. 92, 2931–2946 (2009).
Zhang, Q. et al. Bayesian modeling reveals host genetics associated with rumen microbiota jointly influence methane emission in dairy cows. ISME J. 14, 2019–2033 (2020).
Li, F. et al. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 7, (2019).
Abbas, W. et al. Influence of host genetics in shaping the rumen bacterial community in beef cattle. Sci. Rep. 10, 15101 (2020).
Martinez Boggio, G., Meynadier, A., Buitenhuis, A. J. & Marie-Etancelin, C. Host genetic control on rumen microbiota and its impact on dairy traits in sheep. Genet. Selection Evol. 54, 77 (2022).
Muller, E. E. L. Determining microbial niche breadth in the environment for better ecosystem fate predictions. mSystems. 4, 2379–5077 (2019).
Tapio, M., Fischer, D., Mäntysaari, P. & Tapio, I. Rumen microbiota predicts feed efficiency of primiparous nordic red dairy cows. Microorganisms. 11, 1116 (2023).
Domingues Millen, D. & De Beni Arrigoni, M. & Dias Lauritano Pacheco, R. Rumenology. (2016).
Kruger, B. et al. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 10, 2958–2972 (2016).
Schadt, E. E. et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet. 37, 710–717 (2005).
Chaibub Neto, E. et al. Causal graphical models in systems genetics: A unified framework for joint inference of causal network and genetic architecture for correlated phenotypes. Ann. Appl. Stat. 4, 320–339 (2010).
Pegolo, S. et al. Structural equation modeling for investigating multi-trait genetic architecture of udder health in dairy cattle. Sci. Rep. 10, 7751 (2020).
Rosa, G. J. M. et al. Inferring causal phenotype networks using structural equation models. Genet. Selection Evol. 6, 1–13 (2011).
National Institutes of Health, N. H. G. R. I. A catalog of published genome-wide association studies.
Hu, Z. L., Park, C. A. & Reecy, J. M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 50, D956–D961 (2022).
Acknowledgements
The authors would like to thank all farm staff at the University of Florida Dairy Unit (Alachua, FL, USA) and the Ontario Dairy Research Centre (Elora, ON, Canada), graduate students and interns for all their help with animal handling, sample collection and data acquisition, and Drs. Delaine de Melo and Erika Bonsaglia from the University of California-Davis (Davis, CA, USA) for their help with sample processing.
Funding
This research was supported by the USDA National Institute of Food Agriculture (grant n° 2019-67015-32114) and the Foundation for Food and Agriculture Research (FFAR; grant n° RC109491). Collection of rumen fluid samples, blood samples, and feed efficiency phenotypes in Canadian cows were supported by Dr. Ribeiro’s funding from the Ontario Agri-Food Innovation Alliance (Guelph ON, Canada; project no. 030277) and the Food from Thought Thematic Research Fund (Guelph ON, Canada; project no. 499121). The genotypes of Canadian cows were provided by Dr. Schenkel’s funding from the Dairy Research Cluster 3 (Lactanet, Guelph ON, Canada; and Agriculture and Agri-Food Canada, Ottawa, ON). The funding bodies did not contribute to the design of the study or collection, analysis and interpretation of data and writing the manuscript.
Author information
Authors and Affiliations
Contributions
FP, GJMR, KAW, and GMB conceived and designed the study. HFM, FSL, CCF, RSF, JESP, BM, FSS, and ESR collected and generated the data. GMB analyzed all the data. FP, GJMR, and KAW contributed to the interpretation of the results. GMB wrote the first draft of the manuscript. All authors read, edited, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Martinez-Boggio, G., Monteiro, H.F., Lima, F.S. et al. Revealing host genome–microbiome networks underlying feed efficiency in dairy cows. Sci Rep 14, 26060 (2024). https://doi.org/10.1038/s41598-024-77782-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-77782-z
