
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is a degenerative muscle disease caused by loss of epigenetic silencing and ectopic reactivation of the embryonic double homeobox protein 4 gene (DUX4) in skeletal muscle. The p38 MAP kinase inhibitor losmapimod is currently being tested in FSHD clinical trials due to the finding that p38 inhibition suppresses DUX4 expression in preclinical models. However, the role of p38 in regulating DUX4 at different myogenic stages has not been investigated. We used genetic and pharmacologic tools in FSHD patient-derived myoblasts/myocytes to explore the temporal role of p38 in differentiation-induced DUX4 expression. Deletion of MAPK14/11 or inhibition of p38α/β caused a significant reduction in early differentiation-dependent increases in DUX4 and DUX4 target gene expression. However, in MAPK14/11 knockout cells, there remains a differentiation-associated increase in DUX4 and DUX4 target gene expression later in differentiation. Furthermore, pharmacologic inhibition of p38α/β only partially decreased DUX4 and DUX4 target gene expression in late differentiating myotubes. In xenograft studies, p38α/β inhibition by losmapimod failed to suppress DUX4 target gene expression in late FSHD xenografts. Our results show that while p38 is critical for DUX4 expression during early myogenesis, later in myogenesis a significant level of DUX4 expression is independent of p38α/β activity.
Similar content being viewed by others


Introduction
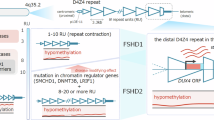
The double homeobox 4 (DUX4) gene, present within D4Z4 macrosatellite repeats, encodes a transcription factor whose function is involved in the coordination of the developmental transcriptional program1. DUX4 acts as an activator for many genes, expressed during the zygotic gene activation, but is subsequently silenced in somatic cells2. In facioscapulohumeral muscular dystrophy (FSHD), silencing is compromised and leads to misexpression of DUX4 in skeletal muscle, causing muscle degeneration3. FSHD type 1 (FSHD1) is caused by contraction of D4Z4 macrosatellite repeats in the subtelomeric region of chromosome 4 (4q35) while FSHD2 results from mutations in epigenetic modifiers such as structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1)4, DNA methyltransferase 3 beta (DNMT3B)5 and ligand dependent nuclear receptor interacting factor 1 (LRIF1)6. Both forms result in epigenetic derepression of 4q35 D4Z4 repeats, presumably allowing access to myogenic transcription factors that promote sporadic expression of DUX4 in rare myonuclei by poorly understood mechanisms3,7.
The pathophysiology of FSHD is incompletely understood partly due to the lack of an animal model that recapitulates regulation of DUX4 from within the primate-specific genomic organization8. Nonetheless, strategies to reduce DUX4 expression are being pursued in the hopes of preserving muscle strength or slowing disease progression. Targeted therapeutic strategies include development of small molecule inhibitors directed towards suppression of DUX4, antisense oligonucleotide9,10,11 and siRNAs12,13,14 targeting DUX4 mRNA which have been validated in preclinical models. Although a range of therapies are being explored, there is a lack of understanding of the myogenic signals that promote DUX4 expression during the complex muscle differentiation process. A more detailed understanding of the factors turning on the DUX4 gene is essential for identifying drug targets for suppressing DUX4 expression in FSHD.
In this context, we and others performed chemical screens to identify small molecules that reduce DUX4 expression. p38 mitogen-activated protein kinase (p38 MAPK) emerged as a druggable target for FSHD15,16. Inhibition of p38α/β MAPK effectively suppressed myotoxic DUX4 expression in cellular and in xenograft animal models of FSHD, paving the way for clinical evaluation of the p38 inhibitor losmapimod in FSHD patients. Indeed, a phase IIb clinical trial of losmapimod (clinicaltrials.gov, NCT04264442) by Fulcrum Therapeutics showed benefits in secondary endpoints (functional outcomes), though it failed to deliver on its primary endpoint (i.e., suppression of DUX4 target gene expression)17. The lack of an observable effect on DUX4 target expression was confounding given the strong preclinical data. It should be noted that previous in vitro and in vivo studies focused on peak expression levels of DUX4, while temporal aspects of DUX4 expression and its regulation by p38 MAPK were not examined. Additionally, single nucleus/single cell level studies on FSHD cells revealed epigenetic memory where DUX4 target gene expression can persist despite absence of DUX4 in some nuclei/cells18,19, underscoring the limits of our understanding.
The above studies indicate that there is need for a comprehensive understanding of the regulation of DUX4 and DUX4 target gene expression in FSHD. With losmapimod entering a phase III clinical trial in FSHD, we still lack an understanding of the molecular mechanisms that link DUX4 expression to differentiating FSHD myocytes and the role of p38 MAPK regulating this process. Here, we focused on characterizing p38-mediated regulation of DUX4 and its target genes during myogenesis in FSHD cells. We observed differential DUX4 expression and regulation in FSHD cells during early (p38-mediated) and late (p38-independent) phases of myogenic differentiation. We noted that during differentiation in vitro, FSHD cells form primary, secondary, and late myotubes with variegated DUX4 expression. Using genetic knockout of p38α/β (MAPK14/11) or pharmacological inhibition, we demonstrate that during primary and secondary myoblast fusion (early differentiation), immortalized FSHD muscle cells exhibit p38-dependent DUX4 bursts. This confirms previous observations15,16 and strongly suggests that p38 MAPK acts as a driver for DUX4 expression during early stages of myogenesis. However, in case of double p38a/β knockouts or pharmacological inhibition of p38a/β, a significant increase in DUX4 expression was observed during late stages of myogenesis in vitro and confirmed in a xenograft model.
Results
FSHD cells exhibit differentiation-induced bursts of DUX4 and DUX4 target genes during myogenic differentiation
To understand temporal regulation of DUX4 by p38, we evaluated DUX4 expression during myogenic differentiation in FSHD patient-derived cells. We first determined if differences existed in the timing of myogenic differentiation between non-FSHD (54−6); D4Z4 contracted FSHD1 (54−2) and D4Z4 noncontracted FSHD2 (MB200) cell lines used in this study. Cells were induced to differentiate and myotubes collected and analyzed for expression of myogenic markers over the course of myogenesis. In non-FSHD and FSHD cells, myogenic differentiation proceeded in three phases, beginning with the formation of primary myotubes (~ 40 h) followed by secondary myotubes (~ 72 h) and late myotubes (> 120 h). We determined the myofusion index (MI) in differentiated cultures based on immunofluorescence staining with anti-MHC. We observed that after 72 and 120 h of differentiation, there were no significant differences in numbers of nuclei per MHC + myotube in FSHD and non-FSHD cells (Fig. 1A, B & Supplementary Fig. 1A). The profiles for the three cell lines were similar with the expected initial rise of the early differentiation marker MYOG (24–48 h) followed by a peak of regeneration marker MYH8 (Schiaffino et al., 2015) (48 h) and a rise in late differentiation marker MYH2 (48 h) which remained elevated with a slight decrease at 96 h before increasing again at 120 h (Fig. 1C). These observations suggest that myogenic differentiation kinetics are similar among non-FSHD and FSHD cell lines in our culture conditions. We next quantified DUX4 and DUX4 target RNA levels in early and late differentiating myotubes using FSHD1 and FSHD2 cells. In early (72 h) myotubes, DUX4 RNA levels are significantly elevated as compared to myoblasts and are further elevated in late myotubes (120 h) (Fig. 1D). A low level of DUX4 target gene expression was present in myoblasts while differentiation induced an increase in DUX4 target gene (ZSCAN4, MDB3L2 and LEUTX) RNA levels that peaked at 72 h (Fig. 1E). At 72 h, a second round of fusion (secondary myotubes) contributed to sustained or increased expression of DUX4 target genes. After this second round of myotube formation, some early myotubes detached from the culture plates coincident with a reduction in DUX4 target gene expression at 96–120 h with exception of LEUTX levels in FSHD2 cells, which were similar between the 72 h and 120 h time points. While DUX4 target RNA levels were generally decreased at later timepoints in comparison to 72 h, they were sustained during late myogenesis at a level much higher than in myoblasts (Fig. 1E). In non-FSHD cells there is no differentiation induced DUX4 target (MBD3L2 and ZSCAN4) gene expression as evidenced in Supplementary Fig. 1B. Further, in undifferentiated FSHD myoblasts, DUX4 target gene expression is significantly higher as compared to non-FSHD myoblasts (Supplementary Fig. 1C).

Induction of DUX4 and DUX4 target genes in FSHD cells during myogenesis. (A) Immunostaining images of MyHC (red) and nuclei (DAPI, blue) on differentiated FSHD1_Unmod and FSHD2_Unmod cells at 72 & 120 h time point. Scale bar: 100 μm, image magnification at 40X. (B) Quantification of MHC + cells from A. MHC + and MHC − nuclei counts were aggregated from three separate imaging fields for each sample. The mean MI values ± Standard Error of the Mean (SEM) were calculated for three samples. Statistical significance was assessed using one-way ANOVA with Dunnett’s post-testing for multiple comparisons. The “ns” indicates that the observed differences were not statistically significant between non-FSHD and FSHD cell lines at the given time points. (C) Relative RNA levels for MYOG, MYH8, and MYH2 at the indicated time points after induction of differentiation in non-FSHD (54−6), FSHD1 (54−2), and FSHD2 (MB200) cell cultures. (D, E) Relative RNA levels for DUX4 (D) and DUX4 targets ZSCAN4, MBD3L2, and LEUTX (E) in differentiating FSHD myocytes at the indicated time points after initiating differentiation. RNA levels were normalized to a housekeeping gene (RPL30), and RNA levels from control FSHD1 myoblasts were used as a reference (set to 1) to calculate relative induction. Error bars represent standard deviation (N = 3), and statistical analysis using one-way ANOVA showed significant differences. **p < 0.001, MT Myotubes.
p38 MAPK drives DUX4 and target gene expression during early myogenesis.
It was previously demonstrated that inhibition of p38α/β with small molecules or knockdown with siRNA significantly suppressed (> 80%) the differentiation-induced increase in expression of DUX4 and its target genes during a 40-h period15 or 5-days on Matri gel plates16. To corroborate those findings, we generated FSHD cells lacking p38α and p38β, individually and in combination, using a CRISPR-Cas9 approach and quantified DUX4 and DUX4 target gene expression at early (72 h) and late (120 h) differentiation time points. Western blot of FSHD cells shows complete loss of p38α (MAPK14) and/or p38β (MAPK11) proteins in respective individual and double knockout (KO) cells (Fig. 2A). RT-qPCR analysis of DUX4 and DUX4 target gene RNA in cells stably expressing Cas9 and “Non-Target” guide was indistinguishable from unmanipulated FSHD cells (data not shown). We used Cas9 as control in all experiments to compare the effect of p38 MAPK KOs on expression of DUX4 and its target genes. In FSHD myoblasts, absence of p38α/β caused a significant reduction in the low level of DUX4 and DUX4 target gene (ZSCAN4, MBD3L2, and LEUTX) RNA levels present in the absence of differentiation signals (Supplementary Fig. 3). The absence of p38α/β also significantly lowered the differentiation-dependent increase in DUX4 and DUX4 target genes expression in comparison to control myotubes at the 72 h time point (Fig. 2B). Complementary to the genetic approach, we treated FSHD cells with losmapimod (1 µM) during differentiation and losmapimod treatment suppressed the expression of DUX4 and its targets during early myogenesis (72 h) ( Fig. 2C), in alignment with previous findings using p38 inhibitors15,16. These data confirm that early myogenic increases in DUX4 RNA levels are largely p38-dependent.

Myogenic induction of DUX4 and DUX4 target genes in FSHD cells with genetic depletion or pharmacological inhibition of p38α/β (MAPK14/11). (A) Deletion of the p38 genes by CRISPR/Cas9 editing in FSHD cells. Western blot analysis of lysates generated from p38 MAPK CRISPR knockouts. Bands for MAPK14 and MAPK11 are shown in parental (Unmodified), Cas9 expressing (Cas9) or genome-edited cell lines with deletion of p38α (MAPK14-/-), p38β (MAPK11-/-) or p38α/β (MAPK14/11-/-). α-Tubulin expression is detected to ensure equal loading. (B) Relative RNA levels for DUX4 and DUX4 targets ZSCAN4, MBD3L2, and LEUTX in differentiating FSHD1 (FSHD1_Cas9), FSHD2 (FSHD2_Cas9) and corresponding double KO lines (_MAPK14/11-/-) at the indicated time points (72 and 120 h) after initiating myogenic differentiation. (C) Relative DUX4 and DUX4 target RNA levels in FSHD1 and FSHD2 myocytes treated with vehicle (_Unmod) or 1 μm losmapimod (_LOS) during a 120-hour differentiation period. Myoblasts in growth media (120 h MB) were compared to differentiated myotubes (120 h MT). (D) Immunostaining images of MyHC (Red) and nuclei (DAPI, blue) on differentiated FSHD1 Cas9 and FSHD1 MAPK14/11-/- cells at 120 h time point with an image magnification at 60X. Scale bar: 100 μm. (E) Quantification of MHC + cells from figure B. MHC + and MHC − nuclei counts were aggregated from three separate imaging fields for each sample. The mean MI values ± Standard Error of the Mean (SEM) were calculated for three samples. Statistical significance was assessed using one-way ANOVA with Dunnett’s post-testing for multiple comparisons. The “ns” indicates that the observed differences were not statistically significant between unmodified and p38α/β double KO FSHD cells (p > 0.05). (F) Immunostaining images of MyHC (Red) and nuclei (DAPI, blue) on differentiated FSHD1 and FSHD2 cells that were treated losmapimod (_LOS) at 120 h time point. Scale bar: 100 μm. (G) Quantification of MHC + cells from figure F. MHC + and MHC − nuclei counts were aggregated from three separate imaging fields for each sample. The mean MI values ± Standard Error of the Mean (SEM) were calculated for three samples. Statistical significance was assessed using one-way ANOVA with Dunnett’s post-testing for multiple comparisons. The “ns” indicates that the observed differences were not statistically significant between untreated and losmapimod-treated FSHD cells (p > 0.05). (H) Relative DUX4 and DUX4 target RNA levels in FSHD1 and FSHD2 myocytes treated with vehicle (_Unmodified_Unmod) or 1 μm losmapimod (_LOS) during a 120-hour differentiation period. Myoblasts in growth media (120 h MB) were compared to differentiated myotubes (120 h MT). In all the qPCR data analysis RNA levels were normalized to housekeeping gene RPL30, followed by respective control groups (considered as 1) and knockout and pharmacological inhibition induced DUX4 and target gene expression fold changes to control were represented.
p38 MAPK-independent DUX4 expression in late differentiating myotubes.
To determine if late myogenic DUX4 expression is regulated by p38 MAPK, we first compared myogenic markers between control and MAPK14/11 double KO cells. We did not observe gross morphological differences in myotubes between control, MAPK14/11 double KOs and losmapimod-treated cells under bright-field microscopic examination in late differentiating cultures (120 h). Further, there was no significant difference in myogenic markers (MYOG and MYH8) as quantified by RT-qPCR (Supplementary Fig. 4A, B). In addition, we measured the myofusion index and observed no significant difference within MHC + multinucleated cells in FSHD Cas9 and MAPK14/11−/− genetic knockout cells (Fig. 2D, E & Supplementary Fig. 4C, D). In line with genetic depletion of MAPK14/11, pharmacological inhibition of p38a/b did not show a significant difference in MI (Fig. 2F, G).
We then measured DUX4 and DUX4 target gene expression in MAPK14/11 double KOs and in losmapimod-treated cells late in myogenic differentiation (120 h). The most striking observation was that there was no significant difference in DUX4 expression between MAPK14/11 double KOs and control cells for both FSHD1 and FSHD2 cell types at the late 120 h time point (Fig. 2C). The expression of DUX4 target genes, however, did not mirror DUX4. In double KO FSHD1 and FSHD2 cells, all three target genes were significantly decreased as compared to control cells (Fig. 2B). In losmapimod-treated cells, DUX4 mRNA levels were significantly elevated in late myotubes as compared to myoblasts although these levels were reduced compared to untreated myotubes in both FSHD1 and FSHD2 cell lines (Fig. 2C). However, in late losmapimod-treated FSHD1 myotubes, the expression of DUX4 target genes was not significantly different from the controls. While DUX4 targets in late FSHD2 myotubes were reduced approximately 50% by losmapimod treatment, their RNA levels were still significantly higher than in myoblasts (Fig. 2H). These observations demonstrate that while p38 MAPK does contribute to DUX4 expression during late myogenesis, there exists a substantial differentiation-dependent increase that is independent of p38α/β activity.
Xenografted FSHD cells express DUX4 and its target genes during early myogenesis while only DUX4 target genes persist later.
Previously, we used a xenograft model for FSHD drug discovery in which FSHD myoblasts were transplanted into injured mouse TA muscles and differentiated in situ. This model was useful to demonstrate that systemic p38 inhibitor treatment suppresses DUX4 expression as transplanted cells differentiate, protects cells from DUX4-mediated toxicity and allows for appropriate muscle differentiation15. In the current study, we profiled gene expression in xenotransplanted FSHD2 myoblasts to determine correlations between differentiation status and DUX4 expression. Figure 3A shows human differentiation marker mRNA levels in whole xenograft TA muscles. The peaks representing early differentiation (MYOG, 4 days), regeneration (MYH3, 8 days) and late differentiation (MYH2, 14 days) confirm successful xenotransplantation (Fig. 3A). A representative micrograph of a 14-day xenograft muscle section is shown in Fig. 3B and demonstrates that a subset set of fibers within a newly regenerated area of the muscle (marked by central nuclei) stain positive for human spectrin (Fig. 3C). As shown in Fig. 3D, DUX4 mRNA levels increased dramatically starting on day 3 after xenotransplantation and peaked on day 4 before declining through day 9. At days 14, 21 and 28, DUX4 mRNA was not detectible in our assays. DUX4 target genes exhibited a similar rise, peaking on days 4 and 5 followed by a gradual decline (Fig. 3E). However, DUX4 target genes remained detectable at a low level (similar to that on day 1) on days 14, 21 and 28 demonstrating persistent expression (Fig. 3E).

DUX4 and DUX4 target expression in FSHD xenograft mice during myogenesis. Muscle xenografts were created by transplanting FSHD2 (MB200) myoblasts to injured TA muscles of immunodeficient mice. At the indicated time points, RNA from excised whole TA muscles was analyzed by qRT-PCR using human-specific primers/probes. (A) Expression profiles of myogenic markers (MYOG, MYH2 and MYH3) in xenografts over four weeks. Values were normalized to housekeeping gene RPL30 and the highest value for each gene was set to one. Error bars represent standard error of the mean. (B) Representative muscle section from a 14-day xenograft TA muscle stained with an antibody specific to human Lamin (brown) with nuclei stained blue. (C) The graph shows the number of human spectrin positive myofibers per section. Error bar represents standard deviation. (D) DUX4 and (E) DUX4 target RNA levels during myogenesis in FSHD xenografts over 28 days. Data is represented as in (A). (F) DUX4 target and (G) regenerative myosin heavy chain (MYH3 and MYH8) RNA levels in mature FSHD xenografts after losmapimod treatment. Losmapimod was administered to xenograft mice orally at 6 mg/kg body weight twice daily for 14 days starting immediately after xenotransplantation. RNA from whole xenograft TA muscles was analyzed by RT-qPCR. Values for each gene were normalized to housekeeping gene RPL30 and data are presented as relative expression with the expression in the vehicle groups set to one. Error bars represent standard error of the mean. ns: non-significant (one-way ANOVA).
Treatment of xenograft mice with losmapimod failed to suppress DUX4 target genes in late myogenic xenografts.
We previously demonstrated that the early differentiation-linked peak of DUX4 expression in the xenograft model (day 4) was suppressed by p38 inhibition15. Although DUX4 mRNA is not detectable past day 9, DUX4 target expression persists at a low level from days 14–28 (Fig. 3E). To determine if this persistent DUX4 target expression is sensitive to p38 inhibition, we treated xenograft mice with losmapimod for an entire 14-day period after xenotransplantation. We observed that expression of DUX4 target genes in mice treated with losmapimod was not significantly different from that in untreated controls on day 14 (Fig. 3F). In the previous study we showed that the late myogenic marker MYH2 levels were unaltered between control and losmapimod-treated group on day 1415. In line with those observations, there were no significant differences in regenerative myogenic markers (MYH8 and MYH3) (Fig. 3G).
Discussion
Myogenic differentiation-linked “bursts” of DUX4 expression are the hallmark of FSHD derived patient muscle cells and the study of this phenomena helped unify our understanding that FSHD is caused by mis expression of DUX4 in adult skeletal muscle20. The discovery that p38 inhibitors could suppress these bursts15,16 led to clinical development of the repurposed p38α/β inhibitor losmapimod for FSHD with confounding Phase IIb trial results. Reports indicated that the trial failed its primary endpoint (i.e., changes in DUX4-driven gene expression) in patient muscle biopsies17. However, several secondary endpoints were sufficiently promising to justify advancing losmapimod to a Phase III trial17,21. The lack of a decrease in DUX4 target expression for patients treated with losmapimod was perplexing considering the robust preclinical data. Technical and biological variables make this a challenging experimental biomarker and difficult to compare to preclinical models. To begin, DUX4 targets are used because DUX4 itself is expressed at a low level in a fraction of nuclei in temporally restricted bursts, defying conventional detection methods in patient biopsies (Beermann et al., 2022). Additionally, biopsies are limited in terms of consistently sampling affected muscle which may contain temporally and spatially diverse microenvironments. Longitudinal muscle imaging studies helped to improve the use of biopsies in FSHD clinical trials (Wong et al., 2020) and led to MRI guided targeting of T2-STIR-positive muscles for sampling. Despite these advancements in clinical trial design, the losmapimod trial still failed to demonstrate DUX4 target suppression. Several mechanisms could explain these results that can be tested in preclinical models of FSHD. One is persistence of DUX4 targets after DUX4 is no longer present. Many DUX4 targets are not expressed in non-FSHD muscle and are robust measures of present or past DUX4 expression, but their temporal relation to DUX4 is complex and they may be present when DUX4 is not18,19,22. Additionally, several reports of non-nuclear DUX4 further complicate the correlation between DUX4 and its target genes23,24. Another potential mechanism is p38-independent DUX4 expression. Here, we demonstrated late differentiation-linked and p38-independent DUX4 expression that occurs after an early differentiation- and p38-dependent increase in DUX4.
We determined the temporal correlation of DUX4 expression with myogenic differentiation of FSHD cells in vitro and in vivo and addressed the relative contribution of p38 MAPK at different myogenic stages. A limitation of our study is that only one FSHD1 and one FSHD2 cell line was used, with the potential for variability in DUX4 and target expression between different FSHD cells25,26. Nonetheless, the cells lines used here gave remarkably similar results. Our observations demonstrated that myogenic differentiation kinetics were similar with some statistical differences at early 24 h and 48 h among non-FSHD and FSHD1 cell lines under our culture conditions, findings in line with Haynes et al.27, where no difference was observed in the expression of myogenic differentiation markers (MYH1, MYH2, and MYOG) between the DUX4-expressing and non-expressing myocytes at the 48 h time point. While DUX4 is known to inhibit myogenesis28,29,30, its induction was not associated with gross alterations of myogenesis and instead coincided with the appearance of early myogenic markers in vitro. In FSHD xenografts, the peaks of DUX4 and DUX4 target expression were coincident with the expression of markers of early differentiation (MYOG) and regeneration (MYH3), suggesting that bursts of DUX4 expression known to coincide with differentiation in vitro31,32,33,34, were also associated with muscle differentiation in vivo. This data suggests that factors driving early myogenesis also promote increased DUX4 expression, in accordance with several studies, e.g26,35, and are consistent with the identification of myogenic enhancers driving DUX4 expression in muscle, although specific transcription factors directly involved have yet to be identified7,36. The activation of p38, known to coincide with and orchestrate early myogenic differentiation37,38,39,40, is consistent with p38 playing a role in DUX4 bursts. Additionally, Brennan et al. employed proteomics to identify phosphorylation changes in response to DUX4 expression and showed that DUX4 itself activated both p38 and JNK kinases, suggesting the existence of a positive feedforward loop between DUX4 expression and p38 activity41. The mechanism by which p38 regulates DUX4 expression likely involves phosphorylation of transcription factors and chromatin regulators that bind to D4Z4 repeats and/or upstream sequences to promote DUX4 transcription. It will be important to identify relevant p38 substrates and determine their temporal contributions to DUX4 expression during myogenic differentiation.
How could p38 inhibitor treatment produce clinical efficacy in-mechanism (i.e. DUX4 suppression) while failing to reduce DUX4 targets in affected muscle biopsies? As mentioned above, one possibility is persistent DUX4 target gene expression after “re-silencing” of DUX4. Another possibility suggested by our data is that DUX4 expression is not solely regulated by the p38 signaling pathway and that p38 inhibition suppresses only the relatively short-lived early differentiation-linked peak of DUX4 expression. We addressed this by evaluating DUX4 expression during early and late phases of myogenic differentiation. It is useful to consider that affected FSHD muscle exhibits a signature of muscle regeneration, marked by the expression of a set of regeneration-related genes including developmental myosin heavy chains42. These genes are expressed early after satellite cell activation and myoblast proliferation when myoblasts fuse to regenerate muscle fibers43. In this context, the key finding of this study was that while p38 drives early myogenic differentiation-linked increases in DUX4, at some point later in differentiation DUX4 expression is not solely regulated by p38. Neither pharmacologic inhibition nor genetic depletion of p38α/β was able to completely prevent differentiation-induced increases in DUX4 in late myotubes in vitro. The pharmacologic consequence of this finding is demonstrated in part using a xenograft model of DUX4 regulation in which FSHD myoblasts transplanted into injured mouse TA muscle differentiate in situ in a semi-synchronous manner. In this model, the expression of regeneration markers (e.g. MYH3) peaks slightly later than DUX4 before slowly declining such that out to more than three weeks, regeneration markers are still expressed at a level above that in myoblasts or in mature muscle fibers. The peak of DUX4 expression during early myogenesis is highly suppressed by p38 inhibitor treatment15; however, DUX4 target expression that persists as the regeneration markers are maintained and late myogenic markers (e.g. MYH2) peak is completely resistant to p38 inhibition. We cannot detect DUX4 mRNA at this late (day 14) time point, although this may be due to limits of our detection assay. Therefore, we cannot conclude directly that p38-independent DUX4 expression occurs in late xenografts. An alternative explanation is that DUX4 targets persist after DUX4 protein declines18,19,22. Nonetheless, the lack of an effect of losmapimod treatment on late xenograft DUX4 target mRNA levels is strikingly similar to reported clinical biomarker results44. The transition from p38-dependent to p38-independent DUX4 expression during myogenesis and its relationship to target gene expression warrants further investigation.
Closing the gap between clinical disease and preclinical models of FSHD has many challenges, a major limitation being our ability to observe and sample the disease state in patients. Since affected muscle biopsies exhibit a regenerating muscle transcription signature42, we speculate they represent a sampling of asynchronously regenerating tissue in which only a small percentage is undergoing “early” differentiation. Therefore, muscle biopsies are unlikely to reveal whether p38 inhibition suppresses early-differentiation and spatiotemporally restricted DUX4 expression peaks. The potential for the process of sampling affected muscle to miss rare regions where DUX4 expression is peaking suggests that DUX4 target gene expression in biopsies may not be a suitable biomarker for therapeutic evaluation of p38 inhibition. Nonetheless, it is important to consider that positive functional outcomes from the Phase IIb trial of losmapimod suggest that suppressing the early differentiation-linked peaks of DUX4 expression has a therapeutic benefit.
Methods
Cell lines
In this study we used previously established isogenic cell lines originally derived from a mosaic patient where the genome has D4Z4 contracted (FSHD1: 54−2 cells) or noncontracted (non-FSHD: 54−6 cells) alleles25. In addition, FSHD2 (MB200) cells harboring a mutation in SMCHD1 were also used for further validation33,45. Myoblasts were grown in Ham’s F-10 Nutrient Mix (Gibco, Waltham, MA) supplemented with 20% USDA–approved source FBS (Corning, Corning, NY), 100 U/100 mg penicillin/streptomycin (Gibco), 10 ng/ml recombinant human fibroblast growth factor (Promega Corporation, Madison, WI), and 1 µM dexamethasone (Sigma-Aldrich, St. Louis, MO) and maintained at 37 °C in a humidified incubator with 5% CO2. For myogenic differentiation, Non-FSHD, FSHD1 and FSHD2 myotubes are harvested after differentiation treatment (DMEM/F12 supplemented with 2% Knockout Serum Replacer (KOSR)), at different time points: 24, 48, 72, 96 and 120 h. In parallel, myoblasts were maintained and collected at different time points (0, 72, and 120 h). All experiments were performed using cell lines between 5 and 20 population doubling (PD) to avoid premature replicative senescence.
Plasmids
Stable Cas9 expressing cells and MAPK14 (p38α), MAPK11 (p38β) knockout cell lines were generated using the following plasmids, all of which were obtained from Addgene: lentiCas9-Blast (Addgene no. 52962), non-target control (Addgene no. 80180), MAPK14 gRNA (Addgene no. 77922; 77923). MAPK11 gRNA was synthesized from vector builder. The sgRNA sequences for MAPK11 are (CCGGGCGTCGTAGGCCGAAC; CCACGCGCGCAGAACGTACC; CCAGTTCGGCCTACGACGCC). Hereafter, in the whole manuscript MAPK14 and MAPK11 will be referred to as p38α and p38β respectively.
Lentivirus production
Lentivirus was produced in HEK293T cells, using standard triple transfection protocols. Briefly, HEK293T were co-transfected with lenti-vector pMDLg/pRRE (Addgene no. 12251); pRSV-Rev (Addgene no. 12253), pMD2.G (Addgene no. 12259) and respective individual plasmids listed above using OptiMEM and Lipofectamine 3000 reagent. After 48 and 72 h, the supernatant was collected and filtered through a 0.45 μm strainer. It was followed by lentivirus concentration, after which the virus titer was calculated, and stored at -80 °C.
Generation of CRISPR/Cas9 MAPK14 and MAPK11 knockout cells
FSHD1 (54 − 2) and FSHD2 (MB200) cells stably expressing Cas9 were generated by transducing cells with Cas9 lentivirus (MOI < 0.3) and selecting with blasticidin (10 µg/ml) for 7 days. Cas9 protein expression was determined by western blot. Further on the technical note, it is very important to use MOI < 0.1 for stably expressing Cas9 in 54 − 2 cells. In 54 − 2 cells, Cas9 itself reduced DUX4 target gene expression at the initial time points (40 h) in comparison to untransduced cells (data not shown), after which there is no difference between control and Cas9 cells especially at 72- and 120 h time points. Stable Cas9 54 − 2 and MB200 cell lines were transduced with MAPK14 and MAPK11 sgRNA lentivirus (MOI < 0.3) in the presence of 2 µg/ml polybrene followed by selection with puromycin (25 µg/ml for 54.2 cells for 7 days; 2 µg/ml for MB200 cells for 2 days), and hygromycin (500 ng/ml) for 7 days for all the cell lines used in this study. Western blot analysis was used to confirm the depletion of p38a/b protein expression. Cells were counted and a limit dilution was performed to obtain a single cell per well culture in a 96-well plate. The best clone was sequenced. After successful clonal selection, knockout (KO) myoblasts were expanded and checked again by western blot for respective protein losses.
Cell culture conditions
Cell cultures from WT, Cas9, non-target control, MAPK14, MAPK11 and MAPK14/11-KOs were seeded at 1 × 105 cells/well in triplicate in 12-well plate. After 2 days in proliferation media, the growth media was removed, and 2 mL of differentiation medium added to induce the formation of multinucleated myotubes. Myoblasts and myotubes were then harvested to assess the influence of MAPK individual and double-KOs on DUX4 and several DUX4 targets (ZSCAN4, MBD3L2, and LEUTX). Myoblasts and myotubes were maintained in parallel and collected at 72 and 120 h. All experimental conditions were tested in triplicate.
Losmapimod treatment
Myoblasts were cultured in standard growth media. One day prior to treatment, 1 × 105 cells/well were plated in 12 well plates. Myoblasts were treated with 1 µM losmapimod (LOS) in growth media. After 48 h of LOS treatment, myoblasts were induced to differentiate into myotubes by replacing growth media with differentiation media supplemented with LOS (1 µM). The induction of differentiation resulted in the formation of primary myotubes at the 48-hour time point. From this 48-hour time point of differentiation onwards, the differentiation culture media with LOS was replaced every 24 h throughout the remainder of the differentiation process. At the specified time point of 120 h, both the differentiating myotubes (treated with LOS) and control myoblasts (not subjected to differentiation) were collected for further analysis along with control myotubes (not treated with LOS). Differentiation media (+/- LOS) and growth media were changed simultaneously as described above to maintain consistency in culture conditions, ensuring that the culture environment remained controlled throughout the myogenic process.
Western blot analysis
To prepare whole-cell lysates, cells were washed twice with cold PBS and lysed in mammalian protein extraction reagent (product No. 78501, Thermo Fisher Scientific) supplemented with protease inhibitor cocktail (product No. A32953, Thermo Fisher Scientific), 2 mmol/L phenylmethylsulfonyl fluoride. Lysates from whole cells were cleared by centrifugation at 12,000 g for 15 min at 4οC, and the supernatants were collected as whole-cell extracts. Protein concentrations were determined using the BCA protein assay kit. Proteins were separated through SDS-PAGE and transferred to nitrocellulose or PVDF membranes which were then blotted with antibodies specific for α-tubulin mouse mAb (926-42213; Li-Cor Biosciences); p38α MAPK polyclonal Rabbit Ab (9218 S; Cell Signaling Technology, Danvers, MA); p38β MAPK (C28C2) Rabbit mAb (2339; Cell Signaling Technology); IRDye 680 goat anti-mouse secondary (926-68070; Li-Cor Biosciences); and IRDye 800 goat anti-rabbit secondary (926-32211; Li-Cor Biosciences). Western blots were then imaged using the LiCor Odyssey CLx and quantified using EmpiriaStudio software.
Real-time PCR (RT-qPCR) analysis
Total RNA was extracted from whole cells using the E.Z.N.A. Total RNA Kit and Tissue RNA Kit (OMEGA Bio-Tek, Norcrass, GA). RNA/DNA was isolated from xenograft tissue using the E.Z.N.A. DNA/RNA kit (Omega Bio-tek, Norcrass, GA). TaqMan Gene Expression Assays (Applied Biosystems) and TaqMan Fast Virus 1-Step Master Mix (Invitrogen) were used for the detection of all genes except DUX4. For DUX4 expression, the isolated RNA was treated with DNase I (Thermo Fisher Scientific) and reverse transcribed into cDNA using Superscript IV (Thermo Fisher Scientific) using only Oligo (dT) primers (Invitrogen) following the manufacturer’s protocol. Detection sequences used in the study were described previously15. Quantitative PCR was performed using a custom TaqMan primer/probe set and TaqMan Gene Expression Master Mix (Applied Biosystems). The relative expression levels of target genes were normalized to that of the reference gene ribosomal protein L30 (RPL30), which was included in multiplex (two gene) PCR reactions, using the ΔΔCt method (Livak and Schmittgen, 2001) after confirming equivalent amplification efficiencies of reference and target molecules. Quantitative real-time polymerase chain reaction (PCR) was carried out on a QuantStudio 5 (Applied Biosystems, Foster City, CA).
Immunofluorescence
FSHD myoblasts (unmodified, Cas9-expressing and MAPK14/11−/− double knockout) were differentiated into myotubes using myogenic differentiation media. In parallel, FSHD cells were treated with losmapimod as described in “Losmapimod treatment” and myofusion index was measured at 72- and 120-hour time points. Cells were fixed in 4% paraformaldehyde for 15 min at room temperature, permeabilized in 2% Triton for 10 min, then washed with PBS. Immunostaining was performed overnight at 4 °C with a 1:250 dilution of MF20 antibody against myosin heavy chain in PBS (+ 0.01% Tween-20,) 1:250 dilution secondary antibody for 1 h at room temperature. Nuclei were counter-stained with DAPI (4′,6-diamidino-2-phenylindole) 1:2000 dilution then washed again. The images were acquired using Keyence BZ-X800 for losmapimod-treated cells and Nikon AX Confocal Microscope System using an immersion lens under 20×-60X magnification for FSHD Cas9 and FSHD MAPK14/11−/− double knockout cells. A fusion index was calculated for each image, where individual nuclei and larger nuclei clusters were segmented. The myofusion index (MI) was defined as the percentage of nuclei found within MHC + multinucleated cells and was quantified for unmodified FSHD, FSHD Cas9 and genetically depleted MAPK FSHD cells or losmapimod-treated cells.
Animals
Male NOD-Rag-IL2gr (NRG) immunodeficient mice (strain #007799 NOD.CgRag1tm1Mom Il2rgtm1Wjl/SzJ; Jackson Laboratories) were used for the xenograft model of FSHD because the absence of T, B, and NK cells in this strain makes them suitable for xenograft transplantation46. Mice were allowed at least 3 days to acclimate to the facility prior to any experimentation. All protocols were approved by the Institutional Animal Care and Use Committee of Saint Louis University. All studies were conducted and reported according to ARRIVE guidelines.
Xenograft studies for monitoring DUX4 expression and regulation.
Immortalized FSHD2 myoblasts were implanted into injured TA muscles and allowed to differentiate in situ. We monitored cell differentiation and the expression of DUX4 and DUX4-induced targets gene over the course of four weeks Briefly, on Day 0 mice are anesthetized with 3–5% isoflurane to effect and 1 × 106 myoblast are administered to the tibialis anterior (TA) muscle in 3 × 10µL injections of cells mixed with 1.2% BaCl2. Mice were euthanized according to ABMA guidelines via CO2 asphyxiation on days 1, 2, 3, 4, 5, 7, 9, 14, 21, and 28. Six mice were analyzed at each time point with the exception of day 4 (n = 12). RNA from whole xenograft TA muscles was analyzed by RT-qPCR using species-specific Taqman primer/probe sets.
Xenograft tissue analysis
For analysis of day 14 FSHD2 xenografts, whole TA muscles were dissected and frozen in 2-methylbutane suspended in a liquid nitrogen bath. Muscle sections were processed by the Saint Louis University Advanced Spatial Biology and Research Histology Facility. Sections were stained with Anti-Lamin A + C antibody [JOL2] ab40567 (Abcam).
Xenograft studies with losmapimod administration
To understand the role of p38 on DUX4 expression during later stages of myogenesis, we xenografted FSHD2 cells to mouse TA muscles as described above and treated the mice with losmapimod for 14 days. Briefly, MB200 cells were prepared and injected into NRG immunodeficient mice as described above. The mice were segregated into 2 groups, Group-1: Vehicle (10% DMSO and 0.5% methylcellulose), N = 12; Group-2: Losmapimod 6 mg/kg BID, N = 12. There were 12 mice in each group at the beginning of the study, 3-mice died due to gavage error in Group 2. Mice were allowed to recover for 1 to 2 h prior to administration of test compound or vehicle which were dosed every 12 h for the duration of the study at a 10 ml/kg dosing volume. The experiment was terminated on day 14. For each in vivo experiment, at the end time point mice were euthanized according to ABMA guidelines via CO2 asphyxiation and tissue samples were collected for RT-qPCR analysis.
Tissue RNA extraction
Entire xenograft TA muscles were harvested, weighed, and homogenized in lysis buffer from kits described above. Lysis buffer was then transferred such that 30 mg of tissue was used in the RNA isolation procedure and 10 mg of tissue was used in the DNA/RNA procedure to prevent column clogging. An additional step was added to digest proteins prior to RNA extraction in both kits. In this step, 300 µl of sample is diluted with 590ul of nuclease free water and 10ul of proteinase K (Omega Bio-tek) and incubated for 10 min at 55º. Samples were centrifuged at max speed for 5 min and supernatant transferred to fresh microfuge tubes. 450 µl of 100% ethanol was added before transferring to RNA columns. The rest of the procedure continued according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed using GraphPad Prism Software. For studies with multiple groups, statistical significance was determined using one-way ANOVA with Dunnett’s posttest, as indicated in the corresponding figure legends. For in vivo data comprising of a single test group versus control and in vitro data a two-tailed t-test was used to determine statistical significance. All samples were normalized to the control and at least three biological replicates per condition were used in all the in vitro studies.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- ANOVA:
-
Analysis of variance
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DNMT3B:
-
DNA methyltransferase 3 beta
- DUX4:
-
Double homeobox 4
- FBS:
-
Fetal bovine serum
- FSHD:
-
Facioscapulohumeral muscular dystrophy
- FSHD1:
-
FSHD type 1
- FSHD2:
-
FSHD type 2
- H:
-
Hour
- H3K4me3:
-
Trimethylation of histone H3 lysine 4
- LEUTX:
-
Leucine twenty homeobox
- LRIF1:
-
Ligand dependent nuclear receptor interacting factor 1
- MAPK:
-
Mitogen-activated protein kinase
- MBD3L2:
-
Methyl-CpG binding domain protein 3 like 2
- RT-qPCR:
-
Reverse transcription quantitative polymerase chain reaction
- siRNA:
-
Small interfering RNA
- SMCHD1:
-
Structural maintenance of 68 chromosomes flexible hinge domain containing 1
- TA:
-
Tibialis anterior
- ZSCAN4:
-
Zinc finger and SCAN domain containing 4
References
Vuoristo, S. et al. <ArticleTitle Language=“En”>DUX4 is a multifunctional factor priming human embryonic genome activation. iScience 25, 104137. https://doi.org/10.1016/j.isci.2022.104137 (2022).
Mocciaro, E., Runfola, V., Ghezzi, P., Pannese, M. & Gabellini, D. DUX4 role in normal physiology and in FSHD muscular dystrophy. Cells 10 https://doi.org/10.3390/cells10123322 (2021).
van der Maarel, S. M., Tawil, R. & Tapscott, S. J. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol. Med. 17, 252–258. https://doi.org/10.1016/j.molmed.2011.01.001 (2011).
Larsen, M. et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur. J. Hum. Genet. 23, 808–816. https://doi.org/10.1038/ejhg.2014.191 (2015).
van den Boogaard, M. L. et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am. J. Hum. Genet. 98, 1020–1029. https://doi.org/10.1016/j.ajhg.2016.03.013 (2016).
Hamanaka, K. et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology 94, e2441–e2447. https://doi.org/10.1212/WNL.0000000000009617 (2020).
Himeda, C. L. et al. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy-associated DUX4 gene. Mol. Cell. Biol. 34, 1942–1955. https://doi.org/10.1128/MCB.00149-14 (2014).
DeSimone, A. M., Cohen, J., Lek, M. & Lek, A. Cellular and animal models for facioscapulohumeral muscular dystrophy. Dis. Model. Mech. 13 https://doi.org/10.1242/dmm.046904 (2020).
Ansseau, E. et al. Antisense oligonucleotides used to target the DUX4 mRNA as therapeutic approaches in faciosScapuloHumeral muscular dystrophy (FSHD). Genes (Basel) 8. https://doi.org/10.3390/genes8030093 (2017).
Lu-Nguyen, N., Malerba, A., Herath, S., Dickson, G. & Popplewell, L. Systemic antisense therapeutics inhibiting DUX4 expression ameliorates FSHD-like pathology in an FSHD mouse model. Hum. Mol. Genet. 30, 1398–1412. https://doi.org/10.1093/hmg/ddab136 (2021).
Lim, K. R. Q. et al. Inhibition of DUX4 expression with antisense LNA gapmers as a therapy for facioscapulohumeral muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A. 117, 16509–16515. https://doi.org/10.1073/pnas.1909649117 (2020).
Vanderplanck, C. et al. The FSHD atrophic myotube phenotype is caused by DUX4 expression. PloS ONE 6, e26820. https://doi.org/10.1371/journal.pone.0026820 (2011).
Lim, J. W. et al. DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum. Mol. Genet. 24, 4817–4828. https://doi.org/10.1093/hmg/ddv206 (2015).
Malecova, B. et al. DUX4 siRNA optimization for the development of an antibody-Oligonucleotide conjugate (AOC) for the treatment of FSHD (P17-13.009). Neurology 98, 1776 (2022).
Oliva, J. et al. Clinically advanced p38 inhibitors suppress DUX4 expression in cellular and animal models of facioscapulohumeral muscular dystrophy. J. Pharmacol. Exp. Ther. 370, 219–230. https://doi.org/10.1124/jpet.119.259663 (2019).
Rojas, L. A. et al. p38alpha regulates expression of DUX4 in a model of facioscapulohumeral muscular dystrophy. J. Pharmacol. Exp. Ther. 374, 489–498. https://doi.org/10.1124/jpet.119.264689 (2020).
Tawil, R. et al. Safety and efficacy of losmapimod in facioscapulohumeral muscular dystrophy (ReDUX4): a randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol. 23, 477–486. https://doi.org/10.1016/S1474-4422(24)00073-5 (2024).
Jiang, S. et al. Single-nucleus RNA-seq identifies divergent populations of FSHD2 myotube nuclei. PLoS Genet. 16, e1008754. https://doi.org/10.1371/journal.pgen.1008754 (2020).
Chau, J. et al. Relationship of DUX4 and target gene expression in FSHD myocytes. Hum. Mutat. 42, 421–433. https://doi.org/10.1002/humu.24171 (2021).
Tawil, R., van der Maarel, S. M. & Tapscott, S. J. Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet. Muscle 4, 12. https://doi.org/10.1186/2044-5040-4-12 (2014).
Wang, L. H. L. T., Han, R., Jiang, J. & Shoskes, J. J. in American Academy of Neurology 2023 Annual Meeting.
Resnick, R. et al. DUX4-Induced histone variants H3.X and H3.Y Mark DUX4 target genes for expression. Cell. Rep. 29, 1812–1820. https://doi.org/10.1016/j.celrep.2019.10.025 (2019). e1815.
Claus, C. et al. The double homeodomain protein DUX4c is associated with regenerating muscle fibers and RNA-binding proteins. Skelet. Muscle 13, 5. https://doi.org/10.1186/s13395-022-00310-y (2023).
Beermann, M. L., Homma, S. & Miller, J. B. Proximity ligation assay to detect DUX4 protein in FSHD1 muscle: a pilot study. BMC Res. Notes 15, 163. https://doi.org/10.1186/s13104-022-06054-8 (2022).
Krom, Y. D. et al. Generation of isogenic D4Z4 contracted and noncontracted immortal muscle cell clones from a mosaic patient: a cellular model for FSHD. Am. J. Pathol. 181, 1387–1401. https://doi.org/10.1016/j.ajpath.2012.07.007 (2012).
Jones, T. I. et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 21, 4419–4430. https://doi.org/10.1093/hmg/dds284 (2012).
Haynes, P., Bomsztyk, K. & Miller, D. G. Sporadic DUX4 expression in FSHD myocytes is associated with incomplete repression by the PRC2 complex and gain of H3K9 acetylation on the contracted D4Z4 allele. Epigenetics Chromatin 11, 47. https://doi.org/10.1186/s13072-018-0215-z (2018).
Bosnakovski, D. et al. Low level DUX4 expression disrupts myogenesis through deregulation of myogenic gene expression. Sci. Rep. 8, 16957. https://doi.org/10.1038/s41598-018-35150-8 (2018).
Bosnakovski, D. et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 27, 2766–2779. https://doi.org/10.1038/emboj.2008.201 (2008).
Young, J. M. et al. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genetics 9, e1003947 (2013). https://doi.org/10.1371/journal.pgen.1003947
Block, G. J. et al. Wnt/beta-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum. Mol. Genet. 22, 4661–4672. https://doi.org/10.1093/hmg/ddt314 (2013).
Rickard, A. M., Petek, L. M. & Miller, D. G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 24, 5901–5914. https://doi.org/10.1093/hmg/ddv315 (2015).
Snider, L. et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 6, e1001181. https://doi.org/10.1371/journal.pgen.1001181 (2010).
Tassin, A. et al. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J. Cell. Mol. Med. 17, 76–89. https://doi.org/10.1111/j.1582-4934.2012.01647.x (2013).
Balog, J. et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics: official J. DNA Methylation Soc. 10, 1133–1142. https://doi.org/10.1080/15592294.2015.1113798 (2015).
Banerji, C. R. S. & Zammit, P. S. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: roles of DUX4 and PAX7. EMBO Mol. Med. 13, e13695. https://doi.org/10.15252/emmm.202013695 (2021).
Bergstrom, D. A. et al. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol. Cell 9, 587–600 (2002).
Penn, B. H., Bergstrom, D. A., Dilworth, F. J., Bengal, E. & Tapscott, S. J. A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 18, 2348–2353. https://doi.org/10.1101/gad.1234304 (2004).
Wu, Z. et al. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol. Cell. Biol. 20, 3951–3964 (2000).
Zetser, A., Gredinger, E. & Bengal, E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J. Biol. Chem. 274, 5193–5200 (1999).
Brennan, C. M. et al. DUX4 expression activates JNK and p38 MAP kinases in myoblasts. Dis. Model. Mech. 15 https://doi.org/10.1242/dmm.049516 (2022).
Banerji, C. R. S., Henderson, D., Tawil, R. N. & Zammit, P. S. Skeletal muscle regeneration in facioscapulohumeral muscular dystrophy is correlated with pathological severity. Hum. Mol. Genet. 29, 2746–2760. https://doi.org/10.1093/hmg/ddaa164 (2020).
Schiaffino, S., Rossi, A. C., Smerdu, V., Leinwand, L. A. & Reggiani, C. Developmental myosins: expression patterns and functional significance. Skelet. Muscle 5, 22. https://doi.org/10.1186/s13395-015-0046-6 (2015).
Fulcrum Therapeutics, I. (2021).
Campbell, A. E. et al. NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. Elife 7 https://doi.org/10.7554/eLife.31023 (2018).
Silva-Barbosa, S. D., Butler-Browne, G. S., Di Santo, J. P. & Mouly, V. Comparative analysis of genetically engineered immunodeficient mouse strains as recipients for human myoblast transplantation. Cell. Transpl. 14, 457–467 (2005).
Acknowledgements
Authors are thankful to Friends of FSH Research, The Chris Carrino Foundation, The FSHD Society and the Muscular Dystrophy Association. We thank Grant Kolar and Caroline Murphy for muscle sample sectioning and IHC.
Funding
This work was supported by a Muscular Dystrophy Association grant to FS and Friends of FSH Research, The Chris Carrino Foundation and FSHD Society grants to RV.
Author information
Authors and Affiliations
Contributions
Conceptualization: AF, FS, JO, RVMethodology: AF, FS, JO, RVInvestigation: AF, FS, JO, RVSupervision: FSWriting—original draft: AF, FSWriting—review & editing: AF, FS, JO, RV.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The animal study protocol was approved by the Saint Louis University Institutional Animal Care and Use Committee (IACUC) for the appropriate care and use of laboratory animals. Saint Louis University is a USDA registered research facility (43-R-011), is regularly inspected and files all required documentation, including an annual report. In addition, under the provisions of the Public Health Service Policy on the Humane Care and Use of Laboratory Animals, the University files required assurance documents to the Office of Laboratory Animal Welfare (OLAW). (OLAW Assurance Number D16-00141). The Animal Care and Use Program at Saint Louis University is also FULLY ACCREDITED by the Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALACi). All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH, Bethesda, Maryland, USA).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vangipurapu, R., Oliva, J., Fox, A. et al. Temporal variation in p38-mediated regulation of DUX4 in facioscapulohumeral muscular dystrophy. Sci Rep 14, 26437 (2024). https://doi.org/10.1038/s41598-024-77911-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-77911-8
Keywords
This article is cited by
-
Diversity challenges and reconciles genetics in facioscapulohumeral muscular dystrophy
Journal of Human Genetics (2025)
