
Abstract
Abnormal growth of blood vessels (choroidal neovascularization) can lead to age-related macular degeneration (AMD) and eventually cause vision loss due to detachment of the retinal pigmented epithelium. This indicates that choroidal neovascularization is important for the treatment of AMD. The circadian clock in the mammalian retina is responsible for controlling various functions of the retina, enabling it to adjust to changes in light and darkness. Recent studies have revealed a potential connection between the circadian clock and eye diseases, although a cause-and-effect relationship has not been definitively established. C57BL/6J male mice (aged 6 weeks) were randomly divided into two groups (Control group: 9:00–21:00 light period (300 lx); Jet lag group: 8-hour phase advance once every 4 days). A laser-induced CNV model was created after 2 weeks of feeding in a controlled or jet-lagged environment. Then, full transcriptome sequencing was performed. The pathways regulated by differentially expressed mRNAs were identified by GO analysis and GSEA. Further protein networks were constructed with the STRING database and Cytoscape software. WGCNA was used to further explore the co-expression modules of these differential genes and the correlation between these differential genes and phenotypes. ceRNA networks were constructed with miRanda and TargetScan. The pathways associated with the overlapping differentially expressed mRNAs in the ceRNA network were identified, and the hub genes were validated by qPCR. A total of 661 important DEGs, 31 differentially expressed miRNAs, 106 differentially expressed lncRNAs and 87 differentially expressed circRNAs were identified. GO and GSEA showed that the upregulated DEGs were mainly involved in reproductive structure development and reproductive system development. The STRING database and Cytoscape were used to determine the protein interaction relationships of these DEGs. WGCNA divided the expression of these genes into several modules and screened the hub genes of each module separately. Furthermore, a ceRNA network was constructed. GO analysis and GSEA showed that these target DEmRNAs mainly function in wound healing, cell spreading, epiboly involved in wound healing, epiboly, and morphogenesis of an epithelial sheet. Finally, ten key genes were identified, and their expression patterns were confirmed by real-time qPCR. In this study, we investigated the regulatory mechanism of ceRNAs in choroidal neovascularization according to different light-dark cycles in the eyeball.
Similar content being viewed by others

Introduction
The retinal pigment epithelial (RPE)-choroid-scleral complex is covered with dense blood vessels. The choroid membrane provides oxygen and nutrients to the outer layer of the retina1. However, under the influence of various complex factors, such as ageing, inflammation, and trauma, abnormal blood vessels develop in the choroid (i.e., choroidal neovascularization). Choroidal neovascularization (CNV) is an important issue in many fundus disorders and is most commonly observed in wet age-related macular degeneration and pathological myopia (primarily in individuals with high myopia (> 600 degrees))2,3. Other conditions include retinal vascular stripes, hereditary macular degeneration, inflammation (such as central exudative chorioretinitis), trauma, tumours, or asymptomatic choroidal neovascularization, all of which may be complicated by CNV4,5. The production of these vessels is also accompanied by an increase in vascular endothelial growth factor (VEGF)6. Through the “driving force” of VEGF, new blood vessels grow like weeds, extending and expanding from the original blood vessel walls. Although the introduction of anti-VEGF drugs has led to significant progress in the treatment of nAMD and improved disease prognosis, this treatment process requires multiple intravitreal injections and only 30–40% of patients have improved vision. Until we gain a better understanding of the primary diseases that cause CNV and their management, current and future treatments will be focused on modifying the course of CNV progression. Exploring the mechanism of CNV is crucial.
The daily rhythms of light/darkness, activity/rest, and feeding/fasting are essential for human physiology. Deviating from these rhythms, such as frequent changes between day and night shifts, increases the risk of disease. Therefore, determination of the impact of disrupted rhythms on human health and how to maintain them for optimal well-being is important. Many diseases are associated with disrupted circadian lifestyles and depend on the pathological deregulation of angiogenesis. Normal angiogenesis is critical for normal tissue function, but when it is disrupted, a range of diseases7,8, including cancer9,10,11, stroke, and heart disease12, can occur. The vascular clock may be regulated differently by central and peripheral circadian clocks in different vasculatures, for example, due to pathological disruption of blood pressure rhythms13, differences in sympathetic innervation14 or expression of receptors for endocrine circadian modulators15, disrupted rhythms of blood sugar levels16,17, or other factors, which may all be important in causing diseases. Therefore, disrupting the circadian clock may impair the physiological regulation of angiogenesis, leading to the development and progression of these diseases. The angiogenic switch is a key regulatory mechanism that controls the balance between angiogenesis and quiescence18. When this switch is activated, endothelial cells lining blood vessels undergo a series of changes that allow them to break free from their supporting cells and migrate towards the site of injury or inflammation. The delicate balance between angiogenesis and quiescence is crucial for maintaining normal tissue function. When this balance is disrupted in favour of angiogenesis, a range of pathological conditions, such as CNV, can occur19,20.
Light is the primary regulators of circadian rhythms21. These cues are detected by nonvisual retinal ganglion cells in the retina, which convey day/night information to the suprachiasmatic nucleus (SCN)22. The disturbance of circadian rhythm can have a series of negative effects on the eyes, but also affect the occurrence and development of related diseases and treatment. However, the effects of circadian rhythm on choroidal neovascularization have not been studied. Therefore, we constructed a circadian disturbance model and revealed a significant increase in choroidal neovascularization after circadian disturbance. In addition, changes in the expression of mRNAs, miRNAs, lncRNAs, and circRNAs in different light rhythms are important for choroidal neovascularization, and the construction of a ceRNA network will help elucidate the molecular mechanism of choroidal neovascularization.
Materials and methods
Ethics statement
The animal studies were approved by the Ethics Committee of Animal Experiments at Nantong University (S20220822-902), and all the processes were in strict accordance with the Standard Operating Procedures for Laboratory Animal Center of NTU. All methods were reported in accordance with the ARVO Animal Statement.
Mouse model of circadian rhythm and jet lag
C57BL/6J male mice (aged 6 weeks) were randomly divided into two groups: circadian rhythm and jet lag, after being acclimated to natural diurnal cycle conditions for one week. The circadian rhythm group was exposed to 300 lx of light period from 9:00 to 21:00 and kept in darkness period from 21:00 to 9:00 to mimic a consistent day-night cycle. The jet lag group experienced an 8-hour phase advance once every 4 days, relative to the normal 12/12-hour cycle. This protocol was followed for an additional three weeks.
Mouse CNV model
C57BL/6 mice were anaesthetized by intraperitoneal injection of 0.1 ml of 0.5% sodium pentobarbital (the anaesthetic dose increased or decreased according to the weight of the mice). Following complete anesthesia, the eyes were dilated using compound tropicamide eye drops. Surface anaesthesia was performed with eye drops of promethazine hydrochloride. After sodium hyaluronate gel was applied to the mouse fundus mirror, laser photocoagulation with a 532 nm frequency doubling (100 μm spot size, 100 MW power, 150 millisecond duration) from the multi-wavelength Krypton ion fundus treatment laser (Coherent, USA) was used at 3, 6, 9, and 12 points, the distance from the optic nerve was approximately 1 optic disc diameter position, four coagulation points were used, and bubble formation was observed.
Fundus fluorescein angiography (FFA)
After the mice were anaesthetized by intraperitoneal injection following laser photocoagulations, 10% sodium fluorescein was diluted to 2% in PBS, and 0.1 ml was injected intraperitoneally. The leakage of fundus angiography was observed with Saris Multi-Modal Ophthalmic Imaging System for Animal (Robotrak, China) 1-minute post-injection. CNV area was quantified in FFA images using ImageJ software (National Institutes of Health, USA). All the data were analysed using GraphPad Prism software (version 9.5.1).
Fluorescence staining of choroidal slides
After the mouse eyeballs were fixed with 4% PFA for 2 h, the cornea, iris, lens and vitreous were removed, and the RPE-choroid-sclera complex was preserved. After permeabilization with 2% Triton and 5% BSA for 30 min, the sections were incubated with an CD31 antibody (R&D Systems, USA) at 4 °C overnight. The samples were removed from the refrigerator the next day and washed three times with PBS for 10 min each time. Next, the tissues were incubated at room temperature for 2 h with the fluorescent secondary antibody of goat anti-rabbit (Abcam, USA). Superfluous liquid was removed from the tissues, and the samples were placed flat on the slide. Avoid light throughout the above process. A DAPI-containing antifluorescence-quenched sealing tablet (SouthernBiotech, USA) was dripped on the slide, and images were taken with Leica Thunder high-resolution fluorescence microscope. Image J software (National Institutes of Health, USA) was used to analyze the fluorescence images. All the data were analysed using GraphPad Prism software (version 9.5.1).
Optical coherence tomography angiography (OCTA)
The mice were anaesthetized with 0.5% sodium pentobarbital by intraperitoneal injection and the eyes were dilated using compound tropicamide eye drops. After ten minutes, the thickness, area and central abnormal blood flow of the lesion were photographed via 400 kHz SS-OCTA instrument (BM400K, TowardPi Medical Technology Co., Ltd., Beijing, China). OCTA data was obtained with a raster scan protocol of 1,536 (horizontal) × 1,280 (vertical) B-scans. We scanned the BOTTOM-RPE layer and observed blood flow imaging in the capillary-free layer of the choroid. The thickness and area were quantified in the OCTA machine. The measurement position was always centered at the fovea without any rotation and the data from the left eyes was horizontally flipped for statistical analysis. Abnormal blood flow areas were quantified using ImageJ software (National Institutes of Health, USA). All the data were analysed using GraphPad Prism software (version 9.5.1).
Western blotting (WB)
RPE-choroid-sclera complexes in mouse eyeballs were extracted, protein extracts were prepared with RIPA buffer (Thermo, USA) containing a protease inhibitor mixture, and protein concentrations were determined with a BCA protein assay (Thermo Fisher Scientific, Massachusetts, USA). After 10% SDS‒polyacrylamide gel electrophoresis, the proteins were transferred to PVDF membranes (Merck, Darmstadt, Germany) and blocked with 5% skim milk, after which the membranes were incubated with primary antibodies at 4 °C overnight (about 16 h) and with secondary antibodies at room temperature for 2 h. Immune complexes were detected using a chemiluminescence kit (SuperSignal ECL Kit, Thermo Fisher, USA) and the mean intensity (intensity/area) of bands was determined and normalized against α-Tubulin using ImageJ software (National Institutes of Health, USA). BMAL1 antibodies were purchased from Cell Signaling Technology, Inc. (1:1000, USA), and other antibodies were purchased from Proteintech (1:1000, China). All the data were analysed using GraphPad Prism (version 9.5.1) software.
RNA isolation, cDNA synthesis, and qRT‒PCR
We isolated total RNA from RPE-choroid-sclera complexes from the control mice and the mice with circadian rhythm disorder with TRIzol reagent (Invitrogen, CA, United States). First-strand cDNA was synthesized from 1 µg of total RNA using a PrimeScript™ RT Master Mix Kit (TaKaRa, Japan). Fast SYBR Premix EX Taq II (TaKaRa, Japan) was used with an ABI Prism 7900 system for qRT‒PCR (Applied Biosystems, Inc., CA, United States). The relative level was calculated by the 2−ΔΔCT relative quantification method. All primer sequences were synthesized by Sangon Biotech (Shanghai, China) and are listed in Supplementary Table 1. All the data were analysed using GraphPad Prism 10 software.
Transcriptome sequencing and data analysis
C57BL/6J male mice (aged 6 weeks) were randomly divided into two groups (Group A: 9:00–21:00 light period (300 lx); Group B: 8-hour phase advance once every 4 days). Laser modelling (approximately 3, 6, 9, and 12 point positions of the optic disc, 100 μm spot size, 100 MW power, and 150-millisecond duration) was performed at 2 weeks for circadian modelling, and RPE-choroid-sclera complexes in eyeballs were harvested after another week of feeding (Fig. 2A). The circadian rhythm disorder group was composed of three mice, and the normal circadian rhythm group was also composed of three mice. After dissecting and extracting the mouse RPE-choroid-sclera complex, a total of 6 samples were obtained from the two groups of tissues and sequenced. The sequencing data have been deposited in the NCBI Gene Expression Omnibus (GEO) database under the accession code GSE253949.

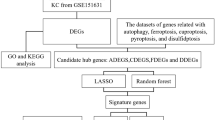
The data analysis process.
Identification of differentially expressed molecules with R software
The differential expression of microRNAs (DEmiRNAs), messenger RNAs (DEmRNAs), long noncoding RNAs (DElncRNAs), and circular RNAs (DEcircRNAs) between the N samples and S samples was identified using the ‘limma’ package. Using the limma R package, we identified DEGs between two risk groups according to the following conditions: log2|FC|>1, an adjusted P‑value < 0.05 and an FDR < 0.05.
Functional annotation and pathway enrichment analysis
To gain a deeper understanding of the functions of DEmRNAs, we performed Gene Ontology (GO) annotation and gene set enrichment analysis (GSEA) using the ‘cluster profiler’ package in R software23. These components provide a comprehensive framework for describing the roles of genes and their products in various biological processes. To further explore the potential pathways in which the DEmRNAs may be involved, we performed GSEA using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) online tool24. Gene Ontology (GO) pathway enrichment analysis was performed on the identified genes. An adjusted P value < 0.05 was used as the cut-off criterion.
PPI network construction and identification of hub genes
Based on the identified DEmRNAs, protein‒protein interaction (PPI) networks were constructed using the Search Tool for the Retrieval of Interacting Genes Database (STRING) (https://cn.string-db.org/) and visualized by Cytoscape 3.9.0. Firstly, miRanda was used to identify the target mRNA, circRNA, and lncRNA of miRNA, with thresholds of SCORE ≥ 150 and ENERGY ≤ − 20. Then calculate the Pearson correlation coefficient (PCC) of the interaction between miRNA circRNA, miRNA mRNA, and miRNA lncRNA. Select miRNA circRNA, miRNA mRNA, and miRNA lncRNA pairs with PCC<-0.8 and p-value < 0.05, and construct a network based on these interaction relationships. When P is less than 0.05, association between two DEmRNAs was considered significant. The confidence scores were set to intermediate values (confidence scores > 0.9). CytoHubba, a plugin for Cytoscape software, was used to identify hub genes. CytoHubba is a plugin for Cytoscape that measures nodes based on their network characteristics, allowing the exploration of important nodes in biological networks25. We obtained the PPI network through STRING and constructed a grid diagram. Default parameters were applied for all plugins to construct sub-networks.
Target gene prediction
The target genes of the DEmiRNAs were identified using miRanda and TargetScan, and the online Venn diagram tool was used to obtain the common mRNAs of the target genes and DEmiRNAs (http://bioinformatics.psb.ugent.be/webtools/Venn/).
WGCNA
Analysis was performed using the R (v4.1.3) package WGCNA (v1.71). After log2 (exp + 1) transformation of TPM expression matrix, the expression matrix of 3167 mRNA significantly differentially expressed genes was extracted, and 2475 genes were left after the genes with too many missing samples or zero variance were eliminated.
Main parameters of the WGCNA (Weighted Correlation Network Analysis (r-project.org)):
type = “unsigned”.
corType = “pearson”.
corFnc = “cor“.
power = 18.
Hubgene Selects parameters: |KME| > 0.8.
Statistical analysis
In this study, all the statistical analyses were conducted using R software (version 4.1.2; https://www.r-project.org/) and GraphPad Prism software (version 9.5.1). Analysis of variance and Student’s t test were used for comparison between groups. A P value < 0.05 was considered to indicate statistical significance. Error bars represent mean ± SEM. ns was not statistically significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Results
Construction of the circadian rhythm mouse model
The data analysis process is illustrated in Fig. 1. The circadian rhythm disorder model is shown in Fig. 2A. We performed laser modelling on all mice during the second week after induction of circadian rhythm disorder. After seven days, we performed fundus coherence tomography angiography (OCTA) for fundus blood flow detection in the mice. As shown in Fig. 2B, clear blood flow was observed at the laser site. Next, we validated the circadian rhythm disorder model using the classic clock gene BMAL1 (Fig. 2C and D), which is the main driver of molecular clocks in mammals. In the modelling group, the expression of BMAL1 was greater than that in the control group, and the expression rhythm was disrupted. The FFA in Fig. 2E shows significant fluorescence leakage at the laser site. The formation of new blood vessels at the laser site determined by immunofluorescence staining for CD31 on the choroidal patch is shown in Fig. 2G. The leakage and area of neovascularization in the circadian rhythm disorder group were more severe than those in the normal circadian rhythm group, as shown in Fig. 2F and H.

The flow of the experiment. (A) Mouse model of CNV. (B) OCTA results of the mice. (C,D) Western blotting of the RPE-choroid-sclera complex. (E,F) The results of fundus fluorescein angiography. (G,H) The results of fluorescence staining of choroidal sections. (n = 3 independent experiments, error bars represent mean ± SEM, ns was not statistically significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Identification of differentially expressed molecules
As shown in Table 1, 282 upregulated mRNAs and 410 downregulated mRNAs were identified between normal samples (N) and jet lag samples (S) using the ‘limma’ package. Among them, 31 genes had different expression patterns in different transcripts. Heatmaps of the top 50 DEmRNAs are shown in Fig. 3A, and volcano plots of all mRNAs are shown in Fig. 3B. A total of 661 DEmRNAs were identified, with 282 upregulated mRNAs and 410 downregulated mRNAs (Fig. 4A); 31 DEmiRNAs were identified, with 27 upregulated miRNAs and 4 downregulated miRNAs (Fig. 4B); 106 DElncRNAs were identified, with 41 upregulated lncRNAs and 65 downregulated lncRNAs (Fig. 4C); 87 DEcircRNAs were identified, with 45 upregulated and 42 downregulated (Fig. 4D); and 15 novel miRNAs were identified, with 10 upregulated and 5 downregulated (Fig. 4E).

The analysis of differentially expressed mRNAs. (A) Heatmap of DEmRNAs. (B) Volcano plot of mRNAs.

The results of different analyses. (A) Heatmap of DEmRNAs. (B) Heatmap of DEmiRNAs. (C) Heatmap of DElncRNAs. (D) Heatmap of DEcircRNAs. (E) Heatmap of DE-novel miRNAs.
The differentially expressed genes included HMOX2, SLC27A, ITGAV, PRESS23, UBl17, APOO, PUM2, UTRN, UBA52, RPL29, SBNO1, ZEB1, SELENOF, CAPZA2, CAPZB, TCOF1, ABHD16A, CTSD, RPL41, SORBS2, DHCR7, PACSIN1, Ml-ATP6, NDUFA10, SGCE, STRA6, OGFOD1, TNNT3, CCND3, RBL2, SRPR, SHISAL1, ZEB2, GTF2I, LRCH3, MAN1A, MT-ND41, RAMP2, PRRC2C, SMARCA2, EVI5, CFH, YWHAG, INPP5A, FAT1, FCHO2, LAMA2, and SNRK. HMOX2 encodes the enzyme haem oxygenase 2, which is an essential enzyme in the haem metabolic process. This enzyme converts haem to biliverdin, which is then converted into bilirubin and carbon monoxide by biliverdin reductase and is related to choroidal neovascularization. Knocking out ZEB1 in tumour endothelial cells inhibited ZEB1/TGF-β/SMAD/VEGFA-ANG2 signalling, which can reduce abnormal angiogenesis through the autocrine pathway, promote the normalization of remaining blood vessels, and increase vascular perfusion26. FAT1 expression is increased in injured arterial tissues of humans and mice, and the inactivation of FAT1 in mouse vascular smooth muscle cells can promote the vascular injury response, significantly increasing the growth of the vascular middle layer and intima. Moreover, FAT1 improves the mitochondrial respiratory function of vascular smooth muscle cells. These studies indicate that FAT1 controls mitochondrial activity to repair and inhibit cell growth, leading to vascular damage27. To assay the quality of samples in each group, principal component analysis was conducted. Based on the sequencing results of all mRNA, miRNA, lncRNA, and circRNA, we revealed the PCA of the two groups through four scatter plots (Supplementary Fig. 1A-D). Similarly, based on the sequencing results of DEmRNA, DEmiRNA, DElncRNA, and DEcircRNA, we revealed the PCA of the two groups using four scatter plots(Supplementary Fig. 1E-H).
GO and GSEA analyses of DEmRNAs
To further investigate the biological roles of these DEmRNAs, we conducted GO analysis and GSEA using the ‘clusterprofiler’ package in R software. The results of these analyses indicated that, in terms of molecular function, the genes were primarily enriched in protein binding, transcription coactivator activity, and collagen binding. In terms of cellular components, the genes were mainly enriched in the Golgi-like trans-Golgi network, Golgi membrane, and Golgi apparatus subcompartments. Finally, in terms of biological process, the genes were predominantly enriched in wound healing, morphogenesis of an epithelial sheet, epiboly involved in wound healing, and epiboly (Fig. 5A). The Golgi apparatus serves as a crucial center for membrane trafficking within mammalian cells. Vascular endothelial growth factor (VEGF) induces the release of vascular endothelial growth factor receptor 2 (VEGFR2) from the Golgi apparatus, and the transportation of VEGFR2 through the Golgi complex is crucial for sustaining the VEGFR2 population on the surface of endothelial cells28. A key activity during the proliferative phase of wound healing is the formation of new blood vessels. Capillary buds infiltrate fibronectin-rich blood clots and gradually develop into a network of tiny blood vessels in the granulation tissue. This network plays a crucial role in supplying essential nutrients and oxygen to the wound bed29. GSEA showed that upregulated DEmRNAs were mainly related to signalling receptor activity, reproductive system development, reproductive structure development, nuclear outer membrane − endoplasmic reticulum membrane network, molecular transducer activity, endoplasmic reticulum subcompartment, endoplasmic reticulum membrane and circulatory system development. In addition, regulation of the mitotic cell cycle phase transition, regulation of the cell cycle phase transition, protein localization to the nucleus, negative regulation of the mitotic cell cycle phase transition, negative regulation of the cell cycle phase transition, myeloid cell differentiation, lipid catabolic processes and haematopoietic or lymphoid organ development were suppressed (Fig. 5B). Activation of the VEGF-receptor pathway triggers a network of signaling processes that promote endothelial cell growth, migration, and survival from pre-existing vasculature30. Lipid metabolism, as a primary form of cellular energy metabolism, significantly influences endothelial cell homeostasis, proliferation, and angiogenesis, holding key pathophysiological implications. The process of angiogenesis involves interactions between growth factors, perivascular cells, the basement membrane, and the extracellular matrix. Endothelial cells (EC) are considered significant contributors to angiogenesis, with key steps including proliferation, migration, sprouting, branching, and tube formation31. The GO and GSEA analysis results indicate that in terms of molecular and cellular functional mechanisms, most differentially expressed genes are enriched in pathways related to neovascularization, which is associated with circadian rhythm disorders.

Pathways enriched in differentially expressed genes. (A) The GO results of the DEmRNAs. (B) GSEA pathways activated and suppressed. (C) Network plot of the PPI results.
Construction of the PPI network of DEmRNAs
To further uncover the potential interactions between proteins encoded by DEmRNAs, using the Search Tool for the Retrieval of Interacting Genes Database (STRING) (https://cn.string-db.org/) and visualized by Cytoscape 3.9.0, we constructed a protein–protein interaction (PPI) network of the DEmRNAs, which included 656 nodes and 208 edges. The PPI enrichment p value was less than 0.00807, and the confidence score was greater than 0.9 (Fig. 5C). The top 10 interaction genes were UBA52, ACTL6A, NDUFV2, CHUK, HIST2H3C2, ATP5C1, NDUFAB1, NDUFB10, TRRAP, SMARCC1 and ribosomal proteins such as RPS14, RPS3A1, RPS21, RPL18, RPS18, RPL11, RPL21, RPL18A, and RPL13A, while some ribosomal proteins related to neovascularization have been reported32. UBA52 identifies as a node with more interaction. It is known as Ubiquitin A-52 Residue Ribosomal Protein Fusion Product 1, is a protein-coding gene which is necessary for DNA repair. It is one of the genes associated with ubiquitin, encoding a fusion protein composed of N-terminal ubiquitin and C-terminal ribosomal protein L4033. Previous studies have shown that UBA52-deficient mice exhibit reduced protein synthesis, cell cycle arrest, and death34. In hepatocellular carcinoma (HCC), UBA52 promotes the proliferation and migration of HCC cells35. One of its related pathways involves the regulation of activated PAK-2p34 through proteasome-mediated degradation and MyD88-dependent cascades initiated on endosomes. ACTL6A is a component of several chromatin remodelling complexes, including SWI/SNF, NuA4/TIP60 histone acetylase, and INO80. The overexpression of ACTL6A has been observed in some cancers36. NDUFV2 contains an iron-sulphur cluster ([2Fe-2 S] binuclear cluster N1a) and is one of the core nuclear-encoded subunits of human mitochondrial complex I37. NDUFAB1 is a mitochondrial acyl carrier protein that acts as a potent cardioprotective agent by increasing the capacity and efficiency of mitochondrial energy metabolism. NDUFB10 is predicted to be involved in the assembly of mitochondrial respiratory chain complex I, which is located in the mitochondrial inner membrane and is expressed in several structures. HIST2H2AC is highly expressed in undifferentiated mammary epithelial cells, and its transcription is activated by growth factors; thus, growth factor-dependent Zeb-1 expression and E-cadherin downregulation are necessary38. ATP5C1 encodes a subunit of mitochondrial ATP synthase, which catalyses ATP synthesis using an electrochemical gradient of protons across the inner membrane during oxidative phosphorylation39,40,41. TRRAP encodes a large multidomain protein of the phosphoinositide 3-kinase-related kinase (PIKK) family and plays a role in transcription and DNA repair by recruiting HAT complexes to chromatin42,43. SMARCC1, an SWI/SNF chromatin remodelling factor, has been reported to play important roles in many cancers.
Hence, with qPCR, our study revealed, for the first time, the roles of 10 hub genes, UBA52, ACTL6A, NDUFV2, CHUK, HIST2H3C2, ATP5C1, NDUFAB1, NDUFB10, TRRAP and SMARCC1, in RPE-choroid-sclera complexes, providing experimental clues for further investigations (Fig. 6). By analyzing the qPCR results, we found that ACTL6A, NDUFV2, and TRRAP were significantly upregulated in the jet lag group, while the other 7 genes were significantly downregulated in the jet lag group.

The expression levels of (A) ACTL6A, (B) ATC5C1, (C) CHUK, (D) HIST2H3C2, (E) NDUFAB1, (F) NDUFB10, (G) NDUFV2, (H) SMARCC1, (I) TRRAP, and (J) UBA52 were determined by RT‒qPCR. The data are presented as the means ± SEMs. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The experiments were repeated four times.
Further, we explored the co-expression modules of these differential genes and the correlation between these differential genes and phenotypes through WGCNA. After finding the optimal soft threshold, the co-expression network was constructed, and the genes were divided into different modules (Fig. 7A-C). Figure 7D shows the correlation between modules, and Fig. 7E shows the correlation between modules and phenotypes. We selected modules with high correlation, and further sorted gene |KME (module eigengene-based connectivity) | > 0.8 in each module (Fig. 7F-M). The first 5 genes of each module were shown in Supplementary Table 2.

WGCNA reveals the co-expression modules of differential genes. (A–B) The optimal soft threshold. (C) The cluster dendrogram shows different modules. (D) The correlation between modules. (E) The correlation between modules and phenotypes. (F–M) The key drivers in interesting modules.
Identified RNA interactions and ceRNA networks
We obtained the targets of miRNAs, lncRNAs and circRNAs via TargetScan and miRanda. The DEmiRNA-DEmRNA network revealed 31 DEmiRNAs targeting 536 DEmRNAs, among which mmu-miR-7033-5p had 311 target DEmRNAs and mmu-miR-1843b-3p had 3 target DEmRNAs (Fig. 8A and B). The network of DEmiRNAs targeting DEmRNAs is shown in Fig. 8D. Among these miRNA targets, several genes play important roles. For example, CPPED1 is a newly discovered serine/threonine protein phosphatase that dephosphorylates AKT1 in the PI3K-AKT signalling pathway44. Deletion of U2AF1 in mouse haematopoietic cells results in severe defects in haematopoiesis, leading to pancytopenia45. TIMP-3 plays a role in VEGF-mediated angiogenesis in vivo in TIMP-3–deficient mice via pathological neovascularization46. According to the DElncRNA Target DEmRNAs network (Fig. 8C), RIP1 is highly expressed in infiltrating macrophages during angiogenesis, and genetic or pharmacological inhibition of RIP1 kinase activity can be effective47. FAM3A is a crucial regulator of vascular constriction and blood pressure48. The inhibition of RUNX1 can significantly inhibit the formation of retinal neovascularization in hypoxia-induced retinal neovascularization model mice49.

The target genes of miRNAs and lncRNAs. (A) The overlapping target DEmRNAs of DEmiRNAs and DEmRNAs. The overlapping DEmRNAs of DElncRNA-trans-DEmRNAs and DEmiRNA targets. (B) The number of target genes of miRNAs. (C) The construction of the ceRNA network. The DElncRNA target DEmRNA network, |corr|>0.9, p value < 0.0001. (D) The DEmiRNA target DEmRNA network, |corr| > 0.9, p value < 0.0001.
The functions of DEmiRNAs targeting DEmRNAs
To explore the regulatory mechanism of the ceRNA network, we performed GO and GSEA analyses. The functions of DEmiRNAs targeting DEmRNA are diverse, such as wound healing, cell spreading, wound healing, signal transduction by p53 class mediators, Ras protein signal transduction, protein mannosylation, morphogenesis of an epithelial sheet, mannosylation, epiboly involved in wound healing, epiboly and the cellular response to viruses (Supplementary Fig. 2A). These genes are located in complex, membrane, actin and so on. The increased pathways include signalling receptor activity, reproductive system development, reproductive structure development, organophosphate metabolic processes, molecular transducer activity, circulatory system development, and cellular response to nitrogen compounds, and the decreased pathways include regulation of the mitotic cell cycle, myeloid cell differentiation and cell cycle phase transition (Supplementary Fig. 2B). These pathways have been reported to be related to neovascularization. The formation of new blood vessels, known as neovascularization, is a complex and multistep process that involves the activation of endothelial cells, degradation of the surrounding basement membrane, formation of tip cells, and sprouting, migration, and proliferation of endothelial cells into the interstitial space. This process is similar to wound healing and requires increased signalling receptor activity for cells to communicate effectively. When a signalling molecule binds to its receptor, it triggers a change inside the cell by altering its shape or activity50. The development of the circulatory system initially occurs through vasculogenesis, the formation of new blood vessels where none previously existed. This process is crucial for neovascularization51.
Discussion
In this study, differentially expressed RNAs in the RPE choroidal sclera complex of mouse eyeballs under different light rhythms were analysed. We identified 661 differentially expressed genes, including HMOX2, ZEB1, FAT1, and SNRK, which play important roles in vasculogenesis. Moreover, 31 differentially expressed RNAs (miRNAs), 100 differentially expressed RNAs (lncRNAs), 87 differentially expressed RNAs (circRNAs), and 15 differentially expressed novel miRNAs were identified.
The pathways affected by these genes include protein binding, transcription coactivator activity, and collagen binding pathways. Choroidal neovascularization requires adequate oxygen and nutrient supplies during development and regeneration, and this process involves new blood vessel formation regulated by proper collagen biosynthesis and crosslinking. Thus, protein binding and protein translation are activated during cellular responses52. These processes are altered in the ribosome, endoplasmic reticulum and Golgi, extracellular exosome and focal adhesion, as collagens are synthesized on ribosomes, processed in the Golgi and endoplasmic reticulum, transported via the extracellular exosome and function in focal adhesion53. According to previous studies, the development of scar tissue can lead to substantial and permanent vision loss. At the cellular level, CNV is a component of several key processes that can be broadly referred to as wound healing or tissue repair. Wound healing involves a coordinated cascade of cellular events driven by the production of cytokines, which are generated by target cells in the context of a continually evolving extracellular matrix (ECM). A similar process occurs in what is clinically termed CNV. Angiogenesis is just one component of this wound healing process. Other key components include inflammation, matrix deposition and remodelling. Thus, in the context of a tissue repair response, viable treatment options for CNV could include therapies other than those that are currently directed at the angiogenic component of this process. For example, anti-inflammatory drugs or antioxidants can be used to reduce inflammation and oxidative stress, which are known to contribute to CNV. Additionally, therapies that target the extracellular matrix, such as matrix metalloproteinase inhibitors or stimulators of matrix deposition and remodelling, could be used to promote the repair of damaged tissue. Furthermore, therapies that target other components of the wound healing process, such as growth factors or adhesion molecules, could also be used to increase the repair response. Notably, these therapies need to be tailored to the specific context of CNV, as each individual’s response to therapy may vary. Additionally, it is crucial to understand the mechanisms by which these therapies exert their effects, as this will help to guide the development of more effective treatments for CNV in the future. GSEA showed that upregulated DEmRNAs were involved in signalling receptor activity, nuclear outer membrane − endoplasmic reticulum membrane network, molecular transducer activity, endoplasmic reticulum subcompartment and endoplasmic reticulum membrane. The regulation of the mitotic cell cycle phase transition, regulation of the cell cycle phase transition, protein localization to the nucleus, negative regulation of the mitotic cell cycle phase transition, negative regulation of the cell cycle phase transition, myeloid cell differentiation through the inhibition of metabolism and cell cycle regulation are essential for the development and differentiation of arterial blood vessels during arterial formation54. The important genes involved in the DEmRNA regulatory network were the UBA52, ACTL6A, NDUFV2, NDUFAB1, and ATP5C1 genes. NDUFV2 is a core nuclear-encoded subunit found in human mitochondrial complex I. NDUFAB1 is a mitochondrial acyl carrier protein that acts as a potent cardioprotector by increasing the capacity and efficiency of mitochondrial energy metabolism. ATP5C1 encodes a subunit of mitochondrial ATP synthase, which utilizes an electrochemical gradient of protons across the inner membrane during oxidative phosphorylation. To verify the reliability of the RNA sequencing data, we performed qRT‒PCR on 10 differentially expressed hub genes (UBA52, ACTL6A, NDUFV2, CHUK, HIST2H3C2, ATP5C1, NDUFAB1, NDUFB10, TRAP, and SMARCC1) in the RPE choroidal sclera complex of mice from the control group and the circadian rhythm disorder group. The experimental results were consistent with the sequencing results.
MicroRNAs (miRNAs) are short noncoding RNAs (ncRNAs) of approximately 22 nucleotides that block mRNA translation or induce mRNA degradation by inducing the Argonaut (AGO) protein to reach the target site of the 3’ untranslated region (UTR) of the mRNA. Many miRNAs are involved in the regulation of vascular growth, remodelling, and inflammation. For example, miR-92a promotes EC dysfunction, inhibits angiogenesis, and promotes atherosclerotic lesions, and antagonists targeting miR-92a have been investigated in phase I clinical studies. Smooth muscle cells (SMCs) are controlled by various miRNAs, including miR-143 and miR-145, whose expression decreases after vascular damage, and their overexpression promotes SMC differentiation and inhibits neointimal formation55. Three different preclinical models of ocular neovascularization (simulated cornea, retina, and choroidal neovascularization, i.e., corneal NV, OIR, and CNV) were used to quantitatively determine whether the miR-29 family (miR-29a/b/c) was significantly downregulated during angiogenesis. Further cell function experiments showed that the miR-29 family had varying effects on the proliferation, migration and lumen formation ability of vascular endothelial cells56. The lncRNA/mRNA coexpression network participates in axonal guidance and mitogen activation of the egg mitogen-activated protein kinase (MAPK) signalling pathway, complement and coagulation cascade reactions, chemokine signalling pathways, and acetone acid metabolism processes. These signalling pathways are associated with angiogenesis, inflammation, and immune disorders, and pathological processes such as neurodegenerative diseases are closely related. For example, the expression of lncRNA-MIAT in retinal pigment epithelial cells treated with high glucose in vitro and in the retina of diabetic rats was significantly greater than that of the controls. Knockout of MIAT alleviates retinal capillary pericellular degeneration and high vascular permeability caused by diabetes, further reducing the viability of vascular endothelial cells in a high-glucose environment in vitro, inhibiting cell proliferation, accelerating cell apoptosis, and blocking tubular formation.
To explore the regulatory mechanism of the ceRNA network involving miRNAs and lncRNAs, we performed GO and GSEA analyses. Targets of DEmiRNAs and DEmRNAs mainly function in wound healing, cell spreading, wound healing, signal transduction by p53 class mediators, Ras protein signal transduction, protein mannosylation, morphogenesis of an epithelial sheet, mannosylation, epiboly involved in wound healing, epiboly and the cellular response to viruses. The increased pathways include signalling receptor activity, reproductive system development, reproductive structure development, organophosphate metabolic processes, molecular transducer activity, circulatory system development, and cellular response to nitrogen compounds, and the decreased pathways include the mitotic cell cycle, myeloid cell differentiation and cell cycle phase transition. These pathways have been reported to be related to neovascularization. The development of new blood vessels, known as neovascularization, is an intricate process that involves the activation of endothelial cells, degradation of the surrounding basement membrane, formation of tip cells, and sprouting, migration, and proliferation of endothelial cells into the interstitial space. This process is similar to wound healing and requires the generation of space in the matrix to allow the formation of a new vessel. Signalling receptor activity is increased, and these pathways are important in cells, as cells normally communicate using signals. When a signalling molecule binds to its receptor, it can cause a change in the shape or activity of the receptor, leading to a cellular response50. The development of the circulatory system begins with vasculogenesis, the formation of new blood vessels where none previously existed. This process is crucial for neovascularization51. Our results revealed that mmu-miR-141-3p can target 98 genes in ceRNAs, as angiogenin promotes angiogenesis via the endonucleolytic decay of miR-14157. Angiopoietin is a family of angio-growth factors that bind to TIE (Tyr kinase with Ig and epidermal growth factor homology domains) receptors. ANG1, ANG2, and ANG4 (human homologues of mouse ANG3) are ligands for TIE2, while TIE1 is an orphan receptor capable of forming heterodimers with TIE2 and regulating TIE2 signalling. The Ang/TIE pathway plays an important role in regulating vascular stability, angiogenesis under physiological and pathological conditions, and inflammation. Among these targets, CD38 has a regulatory effect on vascular remodelling. These target genes are mainly involved in spleen development. The spleen is located on the left side of the abdomen and plays an initial embryonic role in blood formation and later in immune function and red blood cell recycling. The haematopoietic function of the spleen (blood cell formation) is lost with embryo development, and lymphoid precursor cells migrate into the developing organ.
This study also had some shortcomings. On the one hand, more samples are required to verify the stability and reliability of our sequencing analysis results. On the other hand, more in vitro experiments to validate the results of bioinformatics analysis are needed.
Conclusions
This bioinformatic study elucidated the differentially expressed genes, miRNAs, lncRNAs and circRNAs involved in choroidal neovascularization under different rhythmic environments and further elucidated the structure of the ceRNA network. These critical upregulated genes were enriched in signalling receptor activity, reproductive system development, organophosphate metabolic process, molecular transducer activity, circulatory system development, and cellular response to nitrogen compounds, and the downregulated genes were involved in the regulation of the mitotic cell cycle, myeloid cell differentiation and cell cycle phase transition pathways.
Data availability
The original contributions presented in the study are included in the article/Supplementary Material. The sequencing data have been deposited in the NCBI Gene Expression Omnibus (GEO) database under the accession code GSE253949. GEO records will be accessible with the following link after the indicated release date: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE253949. The rest of the data included in the manuscript will be available from the corresponding author on reasonable request.
References
Nickla, D. L. & Wallman, J. The multifunctional choroid. Prog Retin Eye Res. 29(2), 144–168 (2010).
Fernandes, A. R. et al. Exudative versus nonexudative age-related Macular Degeneration: Physiopathology and Treatment options. Int. J. Mol. Sci. 23(5). (2022).
Lu, X. et al. Choroidal Vascular Density Quantification in high myopia with or without Choroidal Neovascularization using Optical Coherence Tomography Angiography. J. Ophthalmol. 2023, 1504834 (2023).
Chen, L. et al. Recognizing atrophy and mixed-type neovascularization in Age-Related Macular Degeneration Via Clinicopathologic correlation. Transl Vis. Sci. Technol. 9(8), 8 (2020).
Gao, R., Ma, J., Zhang, Z., Shang, Q. & Duan, J. Spectral domain-optical coherence tomography retinal biomarkers in choroidal neovascularization of multifocal choroiditis, myopic choroidal neovascularization, and idiopathic choroidal neovascularization. Ann. Med. 53(1), 1270–1278 (2021).
Krause, T. A., Alex, A. F., Engel, D. R., Kurts, C. & Eter, N. VEGF-production by CCR2-dependent macrophages contributes to laser-induced choroidal neovascularization. PLoS One 9(4), e94313 (2014).
Knutsson, A. Health disorders of shift workers. Occup. Med. (Lond) 53(2), 103–108 (2003).
Drake, C. L., Roehrs, T., Richardson, G., Walsh, J. K. & Roth, T. Shift work sleep disorder: prevalence and consequences beyond that of symptomatic day workers. Sleep 27(8), 1453–1462 (2004).
Schernhammer, E. S. et al. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J. Natl. Cancer Inst. 93(20), 1563–1568 (2001).
Kubo, T. et al. Prospective cohort study of the risk of prostate cancer among rotating-shift workers: findings from the Japan collaborative cohort study. Am. J. Epidemiol. 164(6), 549–555 (2006).
Schernhammer, E. S. et al. Night-shift work and risk of colorectal cancer in the nurses’ health study. J. Natl. Cancer Inst. 95(11), 825–828 (2003).
Knutsson, A. Increased risk of ischaemic heart disease in shift workers. Occup. Med. (Lond) 45(1), 55 (1995).
Davidson, A. J., London, B., Block, G. D. & Menaker, M. Cardiovascular tissues contain independent circadian clocks. Clin. Exp. Hypertens. 27(2–3), 307–311 (2005).
Panza, J. A., Epstein, S. E. & Quyyumi, A. A. Circadian variation in vascular tone and its relation to alpha-sympathetic vasoconstrictor activity. N Engl. J. Med. 325(14), 986–990 (1991).
Reilly, D. F., Westgate, E. J. & FitzGerald, G. A. Peripheral circadian clocks in the vasculature. Arterioscler. Thromb. Vasc Biol. 27(8), 1694–1705 (2007).
Su, W. et al. Altered clock gene expression and vascular smooth muscle diurnal contractile variations in type 2 diabetic db/db mice. Am. J. Physiol. Heart Circ. Physiol. 302(3), H621–H633 (2012).
Su, W. et al. Hypertension and disrupted blood pressure circadian rhythm in type 2 diabetic db/db mice. Am. J. Physiol. Heart Circ. Physiol. 295(4), H1634–H1641 (2008).
Jensen, L. D. et al. Zebrafish models to study hypoxia-induced pathological angiogenesis in malignant and nonmalignant diseases. Birth Defects Res. C Embryo Today 93(2), 182–193 (2011).
Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1(1), 27–31 (1995).
Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 9(6), 653–660 (2003).
Fuller, P. M., Lu, J. & Saper, C. B. Differential rescue of light- and food-entrainable circadian rhythms. Science 320(5879), 1074–1077 (2008).
Klein, D. C. & Moore, R. Y. Pineal N-acetyltransferase and hydroxyindole-O-methyltransferase: control by the retinohypothalamic tract and the suprachiasmatic nucleus. Brain Res. 174(2), 245–262 (1979).
Yu, G., Wang Lg Fau - Han, Y., Han, Y., Fau - He, Q-Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. (1557–8100 (Electronic)).
Dennis, G. Jr. Sherman Bt Fau - Hosack DA, Hosack Da Fau - Yang J, Yang J Fau - Gao W, Gao W Fau - Lane HC, Lane Hc Fau - Lempicki RA, Lempicki RA: DAVID: Database for Annotation, Visualization, and Integrated Discovery. (1474-760X (Electronic)).
Chin, C. F., - Chen, S-H. & Chen Sh Fau - Wu, H-H. Wu Hh Fau - Ho C-W, Ho Cw Fau - Ko M-T, Ko Mt Fau - Lin C-Y, Lin CY: cytoHubba: identifying hub objects and sub-networks from complex interactome. (1752 – 0509 (Electronic)).
Fu, R. et al. Inactivation of endothelial ZEB1 impedes tumor progression and sensitizes tumors to conventional therapies. J. Clin. Invest. 130(3), 1252–1270 (2020).
Pastushenko, I. et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 589(7842), 448–455 (2021).
Manickam, V. et al. Regulation of vascular endothelial growth factor receptor 2 trafficking and angiogenesis by Golgi localized t-SNARE syntaxin 6. Blood 117(4), 1425–1435 (2011).
Veith, A. P., Henderson, K., Spencer, A., Sligar, A. D. & Baker, A. B. Therapeutic strategies for enhancing angiogenesis in wound healing. Adv. Drug Deliv Rev. 146, (2019).
Hicklin, D. J. & Ellis, L. M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 23(5), 1011–1027 (2005).
Jiang, Y. et al. 1,6-Hexanediol regulates angiogenesis via suppression of cyclin A1-mediated endothelial function. BMC Biol. 21(1), 75 (2023).
Geng, W., Qin, F., Ren, J., Xiao, S. & Wang, A. Mini-peptide RPL41 attenuated retinal neovascularization by inducing degradation of ATF4 in oxygen-induced retinopathy mice. Exp. Cell. Res. 369(2), 243–250 (2018).
Tiwari, S., Singh, A., Gupta, P. & Singh, S. UBA52 Is Crucial in HSP90 Ubiquitylation and Neurodegenerative Signaling during Early Phase of Parkinson’s Disease. Cells 11(23). (2022).
Kobayashi, M. et al. The ubiquitin hybrid gene UBA52 regulates ubiquitination of ribosome and sustains embryonic development. Sci. Rep. 6, 36780 (2016).
Tong, L. et al. Inhibition of UBA52 induces autophagy via EMC6 to suppress hepatocellular carcinoma tumorigenesis and progression. J. Cell. Mol. Med. 28(6), e18164 (2024).
Yang, Z. et al. ACTL6A protects gastric cancer cells against ferroptosis through induction of glutathione synthesis. Nat. Commun. 14(1), 4193 (2023).
Liu, H-Y., Liao, P-C., Chuang, K-T. & Kao, M-C. Mitochondrial targeting of human NADH dehydrogenase (ubiquinone) flavoprotein 2 (NDUFV2) and its association with early-onset hypertrophic cardiomyopathy and encephalopathy. J. Biomed. Sci. 18(1), 29 (2011).
Monteiro, F. L. et al. The histone H2A isoform Hist2h2ac is a novel regulator of proliferation and epithelial-mesenchymal transition in mammary epithelial and in breast cancer cells. Cancer Lett. 396, 42–52 (2017).
Hayakawa, M. et al. Muscle-specific exonic splicing silencer for exon exclusion in human ATP synthase gamma-subunit pre-mRNA. J. Biol. Chem. 277(9), 6974–6984 (2002).
Matsuda, C., Endo, H., Ohta, S. & Kagawa, Y. Gene structure of human mitochondrial ATP synthase gamma-subunit. Tissue specificity produced by alternative RNA splicing. J. Biol. Chem. 268(33), 24950–24958 (1993).
Pecina, P. et al. Role of the mitochondrial ATP synthase central stalk subunits γ and δ in the activity and assembly of the mammalian enzyme. Biochim. Biophys. Acta Bioenerg 1859(5), 374–381 (2018).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 13(2), 397–406 (2014).
Abbey, D. et al. Lipid droplet screen in human hepatocytes identifies TRRAP as a regulator of cellular triglyceride metabolism. Clin. Transl Sci. 14(4), 1369–1379 (2021).
Haapalainen, A. M., Daddali, R., Hallman, M. & Rämet, M. Human CPPED1 belongs to calcineurin-like metallophosphoesterase superfamily and dephosphorylates PI3K-AKT pathway component PAK4. J. Cell. Mol. Med. 25(13), 6304–6317 (2021).
Dutta, A. et al. U2af1 is required for survival and function of hematopoietic stem/progenitor cells. Leukemia 35(8), 2382–2398 (2021).
Ebrahem, Q. et al. Increased neovascularization in mice lacking tissue inhibitor of metalloproteinases-3. Invest. Ophthalmol. Vis. Sci. 52(9), 6117–6123 (2011).
Ueta, T. et al. RIP1 kinase mediates angiogenesis by modulating macrophages in experimental neovascularization. Proc. Natl. Acad. Sci. U S A 116(47), 23705–23713 (2019).
Xiang, R. et al. VSMC-Specific deletion of FAM3A attenuated Ang II-Promoted Hypertension and Cardiovascular Hypertrophy. Circ. Res. 126(12), 1746–1759 (2020).
Xing, X. et al. RUNX1 can mediate the glucose and O-GlcNAc-driven proliferation and migration of human retinal microvascular endothelial cells. BMJ Open. Diabetes Res. Care 9(1). (2021).
Heldin, C-H., Lu, B., Evans, R. & Gutkind, J. S. Signals and receptors. Cold Spring Harb Perspect. Biol. 8(4), a005900 (2016).
Hillyer, J. F. & Pass, G. The Insect Circulatory System: structure, function, and evolution. Annu. Rev. Entomol. 65, 121–143 (2020).
Smith, M. R. & Costa, G. RNA-binding proteins and translation control in angiogenesis. FEBS J. 289(24), 7788–7809 (2022).
McCaughey, J., Stevenson, N. L., Cross, S. & Stephens, D. J. ER-to-golgi trafficking of procollagen in the absence of large carriers. J. Cell. Biol. 218 (3), 929–948 (2019).
Luo, W. et al. Arterialization requires the timely suppression of cell growth. Nature 589 (7842), 437–441 (2021).
Wang, C. et al. Endothelial-derived extracellular microRNA-92a promotes arterial stiffness by regulating phenotype changes of vascular smooth muscle cells. Sci. Rep. 12 (1), 344 (2022).
Peng, D-W. et al. Anti-angiogenic properties of microRNA-29a in preclinical ocular models. Proc. Natl. Acad. Sci. U S A 119(45), e2204795119 (2022).
Weng, C. et al. Angiogenin promotes angiogenesis via the endonucleolytic decay of miR-141 in colorectal cancer. Mol. Ther. Nucleic Acids 27, 1010–1022 (2022).
Funding
This study was supported by the project of Research and Development of New Technologies and Systems for Low Vision Rehabilitation at Nantong University School of Medicine (23ZH313) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (134422631103).
Author information
Authors and Affiliations
Contributions
YY and SS contributed equally to this work. Study concept and design: SZ and AS; Data collection and processing: YY, JC and XY; Technical and material support: YY and SS; Experiment implementation and date analysis: YY and SZ; Paper writing: YY and SS; Funding support and study supervision: SZ and AS. All authors contributed to the study and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, Y., Su, S., Chen, J. et al. The perspective of ceRNA regulation of circadian rhythm on choroidal neovascularization. Sci Rep 14, 27359 (2024). https://doi.org/10.1038/s41598-024-78479-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-78479-z
