
Abstract
This study was performed for the determination of nineteen azo dyes at trace levels in stream water sample by gas chromatography-mass spectrometry (GC-MS). The selected analytes were preconcentrated with the help of dispersive solid phase extraction (DSPE). Fe3O4 magnetic nanoparticle was modified with covalent organic framework (COF) and the synthesized Fe3O4@COF nanocomposite was used as adsorbent for the separation of the analytes from aqueous solution. The synthesized Fe3O4@COF was introduced to the literature for the first time in this study. Five different parameters (pH, volume of buffer solution, volume of desorption solvent, mixing period and amount of adsorbent) were univariately optimized to acquire high extraction outputs for each analyte. Under the optimum Fe3O4@COF-DSPE-GC-MS conditions, limit of detection (LOD), limit of quantitation (LOQ), linear range (LR), coefficient of determination (R2) and percent relative standard deviation (%RSD) were calculated using external calibration plots belonging to each analyte. LOD values were found between 2.8 and 70.7 μg/kg with corresponding LOQ values ranging between 9.5 and 235.7 μg/kg. In addition, %RSD values were lower than 12.8%. Matrix matching calibration method was used in spiking experiments. Percent recovery results obtained for spiked stream water samples were between 83.1 and 122.4%, with %RSD ≤ 17.5. The high percent recoveries confirmed the accuracy and applicability of the developed Fe3O4@COF-DSPE-GC-MS method for stream water samples.
Similar content being viewed by others
Introduction
Dyes are essential chemicals that give color to paints, textiles, photographs, leather, biological stains, cosmetics and pharmaceutical products, and food. Synthetic dyes broke into the commercial market at the end of the nineteenth century1. Azo dyes are one class of synthetic dyes that have one (monoazo) or several intramolecular N = N bonds2. Azo dyes are commonly used in printing, textile and paper manufacturing, etc3. These chemicals consist of a backbone, the chromophoric groups, the auxochrome groups and the water-soluble groups3,4. Their color depends on the azo bonds, chromophore and auxochrome groups5. Despite their widespread use, most azo dyes have become a threat for human beings due to their non-biodegradable features that lead to skin irritation, allergies, mutagenic, liver, brain, kidney, and central nervous system complications6. The LD50 value for aromatic azo dyes ranges between 100 and 2000 mg/kg b.w7. Some azo dyes are also associated with malignant tumors due to their ability to damage DNA8. Water ecology is also affected by azo dye pollution because of the role they play in inefficient light penetration when present. Additionally, azo dyes can be taken by aquatic organisms and fishes that are in turn consumed by human beings7,9. For these reasons, azo dyes should be accurately and precisely detected in water samples using sensitive analytical methods in order to complement studies regarding their removal from water bodies.
Several analytical instruments such as high-performance liquid chromatography with diode array detection (HPLC-DAD)10, gas-chromatography-mass spectrometry (GC-MS)11, liquid chromatography-mass spectrometry (LC-MS)12, and gel permeation chromatography (GPC)–liquid chromatography–tandem mass spectrometry13 have been used for the quantitation of azo dyes in different samples. GC-MS is a powerful hyphenated technique due to the separation of complex mixtures by GC and molecular identification by MS spectrum14. It is a superior method than LC systems in terms of peak capacity and resolution power15. Hence, the high separation efficiency of GC-MS was used to separate and detect the selected azo dyes.
In the literature, there are several preconcentration methods that are applied before instrumental measurements. These can be classical extraction methods like solid phase extraction (SPE)16 and liquid-liquid extraction (LLE)17, or microextraction methods like liquid-phase microextraction (LPME)18 and solid phase microextraction (SPME)19. Among them, the classical SPE method requires long extraction periods and includes operational difficulties like cartridge clogging20. Another problem with the SPE method is the need for high sample volumes to reach trace level detection limits. In addition, simultaneous extraction of analytes is not easy in the classical SPE method. For these reasons, dispersive solid phase extraction (DSPE) was proposed to the literature to overcome the problems related to SPE application and outcomes. In this extraction method, the adsorbent is dispersed to come into contact with the aqueous solution containing the analyte(s) of interest20. In recent years, several adsorbents have been synthesized and used to separate and preconcentrate analytes in DSPE applications. Fe3O4 nanoparticle21, graphene oxide-SiO2 nanocomposite22, molecular imprinted polymers23, layered double hydroxides24 and metal organic framework25 are some examples of the adsorbents used to extract organic and inorganic species.
Covalent organic frameworks (COFs), firstly produced by Yaghi et al. in 2005, are covalent porous crystalline polymers. These materials have permanent porosity, low mass density and good thermal stability26. COFs can provide π-π interaction and hydrophobic effects when used as adsorbent in SPE methods27. Magnetic nanomaterials can be combined with COFs to provide superparamagnetism, rapid separation and stability28. Fe3O4 is generally used to give magnetic properties to several materials. However, it can be affected by the presence of oxygen in air, which suppresses its magnetism and dispersion in aqueous solutions. For this reason, it is frequently covered with a polymer, metallic or non-metallic substances29,30. Fe3O4@COF adsorbents have been reported in literature for the preconcentration of endocrine-disrupting phenols31, flavonoid glycosides27, peptides32 and copper33.
In this study, a sensitive and selective analytical method for the quantitation of nineteen azo dyes at trace levels in stream water samples was established. The GC-MS system was used to separate and detect the analytes after their preconcentration by the DSPE method. Fe3O4 magnetic nanoparticle was combined with COF to attain magnetic COF nanomaterial for the extraction of the selected azo dyes. To the best of our knowledge, the Fe3O4@COF adsorbent was synthesized and utilized as adsorbent for the first time in this study. Significant experimental parameters such as adsorbent dosage, pH, buffer solution volume, eluent type and volume, vortex period were individually optimized to enhance the signal-to-noise ratio for each analyte. After the optimization experiments, system analytical performance was evaluated in terms of limit of detection, limit of quantitation, and linear range. Accuracy and applicability were checked by spiking experiments carried out on stream water samples.
Materials and methods
Chemicals and reagents
Analytical grade chemicals and reagents were used in all experimental studies. Azo dye mixture (300 μg/mL in acetonitrile) was obtained from Chem Service Inc. (West Chester, USA). Acetonitrile (≥ 99.9%), ethanol (≥ 99.9%), methanol (≥ 99.9%), dimethylsulfoxide (≥ 99.9%), tetrahydrofuran (≥ 99.5%), acetic acid (≥ 99.8%), ammonia (25.0–30.0%), FeCl3.6H2O (99.0-102.0%) and Fe(NH4)2(SO4)2.6H2O (99.0-101.5%) were purchased from Merck (Darmstadt, Germany). Terephthalaldehyde (99%) and tris(4-aminophenyl)amine (97%) were obtained from Sigma Aldrich (Darmstadt, Germany). Ultrapure water was obtained from an Elga Pure Flex 3 Ultrapure Water Treatment System (High Wycombe, United Kingdom).
Instrumentation
Chromatographic separation of the selected analytes was performed with a 6890 model Agilent Gas Chromatograph, and mass spectra of the analytes was obtained by an Agilent Mass Spectrometer. Helium at a flow rate of 1.8 mL/min with a constant linear velocity was used as carrier gas and (5%-phenyl)-methylpolysiloxane phase capillary column (30 m; 250 μm; 0.25 μm) was used as stationary phase. Sample injection volume, injection port temperature and injection mode were 1.0 μL, 280 °C and splitless, respectively. The oven temperature program employed consisted of an initial ramp from 70 to 130 °C (held for 1.0 min) at a rate of 17 °C/min, and the second ramp from 130 to 290 °C (held for 2.27 min) at a rate of 50 °C/min. MS transfer line, MS source and MS quad temperatures were 280 °C, 230 °C and 150 °C, respectively, with an ionization voltage of 70 eV and a full scan mass range of m/z 40–600.
Characterization of the Fe3O4@COF was performed using Fourier Transform infrared spectrometer (Thermo Scientific Nicolet IS10 model, Massachusetts, USA), scanning electron microscope (Zeiss EVO LS10 model, Oberkochen, Germany) and X-ray diffractometer (PANalytical X′Pert Pro model, Malvern, UK).
Fe3O4@COF synthesis
Fe3O4 was synthesized according to a study reported in the literature34. Firstly, Fe3O4 nanoparticle was synthesized using 8.1 g of FeCl3.6H2O and 5.9 g of Fe(NH4)2(SO4)2.6H2O dissolved in 200 mL ultrapure water. The iron-containing mixture was heated to 80 °C with vigorous stirring. Next, 20 mL of concentrated NH4OH (%25, w/v) was added into the iron-containing solution in a dropwise manner. The reaction was kept at 80 °C for 2.0 h under nitrogen gas atmosphere. After the completion of the reaction, the mixture was allowed to cool to room temperature and the obtained black product was separated with the help of a neodymium magnet. Ultrapure water and ethanol were used to wash the black product before drying at 50 °C in a laboratory oven.
In the second part of the synthesis procedure, modified from a study in literature32, 61.2 mg terephthalaldehyde, 86.7 mg tris(4-aminophenyl)amine and 157.7 mg Fe3O4 nanoparticle were weighed and put into an Erlenmeyer flask. Approximately 60 mL of dry dimethylsulfoxide was pipetted into the flask. After the solution was sonicated for 20 min, 2.0 mL of concentrated acetic acid solution was added to the solution. The reaction was completed after an additional sonication process for 40 min. The dark red product was washed with 60 mL dry tetrahydrofuran, 60 mL dry methanol and 60 mL dry acetonitrile. The product was dried at 50 °C for 48 h. The synthesis procedure for Fe3O4@COF is given in Fig. 1. In addition, COF without Fe3O4 was also synthesized according to the above procedure without the addition of Fe3O4.

Synthesis of COF using terephthalaldehyde and tris(4-aminophenyl)amine.
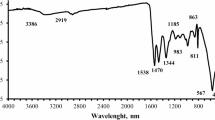
Fourier transform infrared spectrometry (FTIR) was used to characterize the synthesized Fe3O4@COF composite. According to Fig. 2, the absorption band at 548.17 cm− 1 can be attributed to Fe-O-Fe vibration. The absorption band at 1696.57 cm− 1 is ascribed to C = N vibration, which proved the successful synthesis of the Fe3O4@COF composite. The other peaks at 1615.59, 1492.15, 1190.83 and 830.21 cm− 1 are assigned as C = C stretching, C-C stretching in an aromatic ring, C-N stretching and 1,4-disubstituted aromatic C-H bending.

FTIR spectrum for Fe3O4 and Fe3O4@COF nanocomposite.
The shape, size and surface morphology of the synthesized Fe3O4@COF material were checked using SEM. According to Fig. 3 (A, B), COF showed uniform, solid and spherical morphology. Figure 3 (C, D) indicated that the Fe3O4@COF material had nano-sized structures and a homogenous size distribution with a texture composed of unevenly spherical particles. It had a rough surface after the combination of Fe3O4 and COF. The SEM images proved the modification of Fe3O4 with COF. In addition, one experiment was also performed to check the success of the Fe3O4@COF synthesis. Magnetic field was applied to the liquid filtrate obtained after the washing step of the product. There were no solid particles left in the filtrate. This experiment proved that Fe3O4 was successfully modified with COF to generate the magnetic COF adsorbent.

SEM images for the synthesized COF at 200 nm scale (A) and 1.0 μm scale (B) and Fe3O4@COF at 200 nm scale (C) and 1.0 μm scale (D).
The XRD pattern of the prepared Fe3O4@COF nanocomposite is given in Fig. 4. The XRD spectrum had the prominent peaks at 30.26°, 35.55°, 35.73°, 43.35°, 53.69°, 57.24°, 62.70° and 63.10° 2θ angles for the synthesized Fe3O4@COF nanocomposite. These peaks were in agreement with literature findings31,32,35. The size of the Fe3O4@COF nanocomposite was calculated with the aid of Debye-Scherrer equation36. According to the calculations, the nanocomposite had an average particle size of 43.6 ± 2.5 nm in terms of the three most intensive peaks (35.55o, 35.73o and 63.10o).

XRD spectrum for the synthesized Fe3O4@COF nanocomposite.
Fe3O4@COF-DSPE procedure
Approximately 8.0 mL of aqueous standard or sample solution and 1.50 mL of pH 7.0 buffer solution were added to a clean 15 mL centrifuge tube containing pre-weighed 10 mg Fe3O4@COF adsorbent. Mixing by vortex was applied to the mixture for 30 s followed by centrifugation for 2.0 min to ease the decantation process. A neodymium magnet was used to collect the adsorbent at the bottom of the tube during the decantation process. In the final step, 75 μL acetonitrile was added to desorb the analytes from the adsorbent. The analyte rich phase was injected into the GC-MS system.
Samples
Three stream water samples were collected from different regions of Türkiye. All stream water samples were filtrated using 0.45 μm syringe filters to remove the solid particles from the samples. In recovery experiments, 1.40 g stream water sample was diluted to 35 g with ultrapure water after the sample was spiked to five different concentration levels. Acetonitrile concentration in all spiked solutions was fixed to %1.0 (w/w) in order to abstain from different organic solvent content in the solutions that may affect the adsorption of the analytes onto the adsorbent. The developed Fe3O4@COF-DSPE method was applied to the spiked stream water samples.
Results and discussion
The Fe3O4@COF based DSPE method was developed for the simultaneous preconcentration of nineteen azo dyes in stream water samples. Parameters including pH, buffer solution volume, desorption solvent volume, mixing period and dosage of adsorbent affecting the extraction efficiency were individually examined to optimize the DSPE process. The peak areas of the nineteen azo dyes were used to assess the parameters and define optimum conditions. Each set of experiments was repeated three times in order to calculate standard deviation and percent relative standard deviation (%RSD).
Effect of type and volume of buffer solution
Due to its ability to change the ionic state of analytes and the functional groups on adsorbents, pH is a crucial determinant in both adsorption and desorption procedures37. Additionally, the extraction efficiency of the selected analytes on the adsorbent could be enhanced using an appropriate pH value38. Therefore, the effect of pH on the extraction of the selected azo dyes was evaluated in the range of pH 3.0–8.0, using 1.0 mL of a suitable buffer solution. As shown in Fig. 5, it was observed that the azo dyes did not record sufficient extraction efficiency in the acidic medium due to weak protonation. This may cause a decrease in the hydrophobic and π-π interactions between Fe3O4@COF and azo dyes39. pH 7.0 was recorded as the most effective value for ten analytes. In addition, analytes with low signal-to-noise ratios had the highest peak area values at pH 7.0. For this reason, the pH of the initial sample solution was selected as 7.0, which exhibits high extraction outcomes for most of the analytes studied.

Optimization of pH value. (1: 4-aminobiphenyl, 2: Benzidine, 3: 4-chloro-2-methylaniline, 4: 2-naphthylamine, 5: o-aminoazotoluene, 6: 2-amino-4-nitrotoluene, 7: 4-chloroaniline, 8: 4,4’-methylenedianiline, 9: 3.3’-dichlorobenzidine, 10: 3,3’-dimethylbenzidine, 11: 4,4’-diamino-3,3’-dimethyldiphenylmethane, 12: 4,4’-oxydianiline, 13: 4,4’-thiodianaline, 14: 2,4,5-trimethylaniline, 15: o-anisidine, 16: 3,3’-dimethoxybenzidine, 17: p-cresidine, 18: 4,4’-dimethylene bis (2-chloroaniline), 19: 2,4-dimethylaniline).
Subsequently, four different volumes of the pH 7.0 buffer solution (0.50, 1.0, 1.5, and 2.0 mL) were tested to find the optimum value. According to the result, most analytes (14 out of 19 analytes) showed higher extraction efficiencies for the 1.5 mL volume of buffer solution. Hence, 1.5 mL of pH 7.0 was selected as optimum value for further optimization steps.
Effect of desorption solvent volume
The volume of desorption solvent is one of the essential parameters to achieve accurate and repeatable results for analytes38. The desorption of analytes was carried out using acetonitrile, which is an appropriate solvent for the analytes. For this purpose, the effect of acetonitrile volume was studied using different volumes (75, 100, 150, and 200 μL) to ascertain the desorption capability. The experimental results showed that peak areas gradually increased with the decrement of acetonitrile volume due to less dilution of the analytes. The highest peak area for seventeen out of nineteen analytes was recorded as 75 μL of acetonitrile. The increase in the first/last sample volume ratio is important for the acceptable preconcentration factor and extraction efficiency. As a result, 75 μL of acetonitrile was chosen as the optimum desorption solvent volume for the next optimization study.
Effect of vortex period
In this study, vortex was used to mix the analyte solution containing the adsorbent. Its period should be optimized in order to reach high extraction outputs and adsorption equilibrium40. Extraction trials for vortex period were performed at intervals of 0.0, 15, 30, and 60 s to examine the impact of the vortex period on extraction efficiency. For most of the analytes, low peak areas were obtained for the experiments performed without mixing. It demonstrates that the analyte and adsorbent did not have sufficient interaction in the absence of the mixing step. For most of the analytes (seventeen out of nineteen analytes), peak area values increased with the increment of the vortex period. Further, there was no notable change in both peak areas and standard deviation between 30 and 60 s. Therefore, 30 s was determined as the optimum vortex period to achieve quick and efficient extraction of the analytes.
Effect of adsorbent dosage
Fe3O4@COF adsorbent has advantages such as highly efficient adsorption capacity with fast separation performance owing to its magnetic core28. For this reason, the optimization of Fe3O4@COF dosage is one of the key parameters in the separation/extraction step of the analytes. The effects of adsorbent dosage on the extraction efficiency of the selected analytes were tested by trying 0, 5.0, 10, 15, and 20 mg of Fe3O4@COF adsorbent. As seen in Fig. 6, the highest and repeatable results for 11 analytes were obtained for 10 mg. In addition, 5.0 mg was not sufficient to collect the analytes. Hence, 10 mg of Fe3O4@COF adsorbent was adopted as the optimum adsorbent dosage.

Optimization of adsorbent dosage. (1: 4-aminobiphenyl, 2: Benzidine, 3: 4-chloro-2-methylaniline, 4: 2-naphthylamine, 5: o-aminoazotoluene, 6: 2-amino-4-nitrotoluene, 7: 4-chloroaniline, 8: 4,4’-methylenedianiline, 9: 3.3’-dichlorobenzidine, 10: 3,3’-dimethylbenzidine, 11: 4,4’-diamino-3,3’-dimethyldiphenylmethane, 12: 4,4’-oxydianiline, 13: 4,4’-thiodianaline, 14: 2,4,5-trimethylaniline, 15: o-anisidine, 16: 3,3’-dimethoxybenzidine, 17: p-cresidine, 18: 4,4’-dimethylene bis (2-chloroaniline), 19: 2,4-dimethylaniline).
System analytical performance of Fe3O4@COF-DSPE-GC-MS system
The developed method was investigated in terms of system analytical performance parameters including limit of detection, limit of quantitation, linear range and coefficient of determination. A series of standard solutions prepared between 6.5 and 3965.7 μg/kg were gravimetrically prepared using three different stock solutions in acetonitrile. Acetonitrile concentration was fixed to 1.0% in all standard solutions. External calibration plots were drawn for each azo dye compound and the LOD, LOQ, LR, and R2 values recorded are given in Table 1. The LOD values for the analytes ranged from 2.8 to 70.7 μg/kg (mass based). Based on the information given by Magnusson B. in the literature41, precision is assessed by repetitive measurements of the lowest concentration in the calibration plot of each analyte. %RSD values for the lowest concentrations in calibration plots were calculated between 6.2 and 12.8%.
Ion-pairing high performance liquid chromatography tandem mass spectrometry (LC-MS/MS)42, microextraction by packed sorbent-gas chromatography-mass spectrometry (MEPS-GC-MS)43, salting-out assisted liquid–liquid extraction gas chromatography-mass spectrometry (SALLE-GC-MS)43, solid phase microextraction- gas chromatography-mass spectrometry (SPME-GC-MS)44 are some analytical methods used for the preconcentration and determination of several azo dyes. According to analytical performance parameters given in Table 2, the developed Fe3O4@COF-DSPE-GC-MS method had wider LR values than the other methods. LOD and LOQ ranges of the developed methods were higher or similar to the other preconcentration methods. However, other methods consisted of experimental procedures with multiple extraction steps and additional chemicals. The analytical method developed by Tölgyesi et al.42 needed an ion-pairing reagent (heptafluorobutyric acid) and LC-MS/MS system that has high operational cost and organic solvent usage. The other study used a special automated system for the MEPS method, drying process of the sorbent and washing process of the cartridge, which can be time-consuming for an analytical procedure. In the same study, the SALLE method needed 750 μL ethyl acetate and chemical reduction including sodium dithionite, which is toxic and hazardous for health and the environment. Cioni et al.44 presented an analytical method including chemical reduction with sodium dithionite and SPME fiber. It is known that SPME fibers are more fragile45 and chemical reduction includes complex procedures and pH adjustments46. It can be concluded that the developed Fe3O4@COF-DSPE-GC-MS can be used to obtain trace levels of the selected analytes with short extraction periods and less chemical usage. When the developed method was compared to other solid phase-based extraction/microextraction methods, the magnetic property of the Fe3O4@COF adsorbent facilitated easier adsorbent separation and faster extraction procedure for the preconcentration of azo dyes.
Recovery experiments
The developed Fe3O4@COF-DSPE-GC-MS method was examined in terms of accuracy and applicability. For this purpose, three different stream water samples were spiked at five concentration levels in the range of 100 and 600 μg/kg. The spiking procedure is detailed in the “Samples” section. Each sample was preconcentrated by the developed Fe3O4@COF-DSPE method and calibration plots for analytes were obtained. Matrix matching calibration method has been commonly used in literature to minimize positive and negative effects on analyte signals caused by sample matrices. The standard solutions used to draw the calibration plot are adjusted to match the sample matrix47. Matrix matching calibration strategy was carried out to take into account the sample matrix effect. Percent recovery results for Sample Z and B were found using the linear equations obtained for Sample K. Tables 3 and 4 summarize all percent recovery results with their %RSD values for Sample Z and B. Percent recoveries for the spiked Sample Z ranged from 83.9 to 110.8%, while that for the spiked Sample B varied between 83.1 and 122.4%. According to the calibration plots obtained by external and matrix matching calibrations, the slope of the plots were significantly different from each other, meaning that there was matrix effects for each analyte. For example, 4-aminobiphenyl had a slope of 9707.1 for the external calibration plot and 42225.08 for the matrix matching calibration of Sample K.
Greenness and practicality of the developed method
Anastas and Warner identified twelve principles of green chemistry that enhance experimental procedures for the protection of human health and the environment48. In 2012, an eco-scale analytical tool was proposed to evaluate the greenness of the procedures by giving penalty points to chemicals, reagents and instruments used in the procedures. In this tool, a procedure has 100 points in the beginning and all penalty points are subtracted from 100 points49,50. According to the greenness evaluation proposed by Gałuszka et al.49, the developed method can be categorized as acceptable green due to the analytical eco scale point of 69, which was calculated by subtracting all penalty points for the synthesis and extraction. In addition, the practicality of the method was checked by blue applicability grade index (BAGI) software. As can be seen in Figs. 7 and 65 points were attained for the developed method, and it can be concluded that the method was practical based on the BAGI software, indicating that an analytical method is practical if it has at least 60 points51.

Pictograms obtained by BAGI software.
Conclusion
In the presented work, a sensitive, simple and fast analytical method was established for the determination of nineteen azo dyes at trace levels in stream water samples. The DSPE method was used in the preconcentration step and the GC-MS system was operated as the instrumental separation and detection method. The novelties of this study are the adsorbent, which was synthesized and reported for the first time in literature, and the application of the adsorbent for the preconcentration of the selected azo dyes from water samples. The Fe3O4@COF adsorbent was synthesized using Fe3O4 nanoparticles as the magnetic core and terephthalaldehyde and tris(4-aminophenyl)amine as the organic ligand. Under the optimum DSPE conditions, system analytical performance parameters were determined for each azo dye. Trace level LOD and LOQ values were attained thanks to the developed preconcentration method. Spiking experiments were carried out on stream water samples to validate the accuracy and applicability of the developed method. The proposed preconcentration method prior to GC-MS analysis exhibited wide linear range, good sensitivity and easy operation. It can be concluded that the proposed method is suitable and applicable to other azo dye compounds in similar sample matrices.
Data availability
Data will be made available on reasonable request. Kindly contact with the corresponding author.
References
Nikfar, S. & Jaberidoost, M. Dyes and colorants. in Encyclopedia of Toxicology 252–261 (Elsevier, https://doi.org/10.1016/B978-0-12-386454-3.00602-3. (2014).
Le Coz, C. J. & Dyes In Encyclopedia of Toxicology104–114 (Elsevier, 2005). https://doi.org/10.1016/B0-12-369400-0/00359-8
Benkhaya, S., M’rabet, S. & El Harfi, A. Classifications, properties, recent synthesis and applications of azo dyes. Heliyon 6, e03271 (2020).
Al-Rubaie, L. A. A. R. & Mhessn, R. J. Synthesis and Characterization of Azo Dye Para Red and new derivatives. E-J. Chem. 9, 465–470 (2012).
Gürses, A., Açıkyıldız, M., Güneş, K. & Gürses, M. S. Classification of Dye and Pigmentsin 31–45 (Springer, 2016). https://doi.org/10.1007/978-3-319-33892-7_3
Hijazi, A. et al. Biosorption of methylene blue from waste water using Lebanese Cymbopogon citratus (citronnelle). Eur. Sci. J. 11, (2015).
Okeke, E. S. et al. Analytical detection methods for azo dyes: a focus on comparative limitations and prospects of bio-sensing and electrochemical nano-detection. J. Food Compos. Anal. 114, 104778 (2022).
Mahbub, K. R., Morium, B., Ahmed, M. M., Akond, M. A. & Andrews, S. Decolourization of Novacron Blue and Novacron Super Black Azo Dyes by Bacillus spp isolated from Textile Effluents in Bangladesh. J. Sci. Res. 7, 45–53 (2015).
Sen, S. K., Raut, S., Bandyopadhyay, P. & Raut, S. Fungal decolouration and degradation of azo dyes: a review. Fungal Biol. Rev. 30, 112–133 (2016).
Dong, M. Y., Wu, H. L., Long, W. J., Wang, T. & Yu, R. Q. Simultaneous and rapid screening and determination of twelve azo dyes illegally added into food products by using chemometrics-assisted HPLC-DAD strategy. Microchem J. 171, 106775 (2021).
Balçık, U., Chormey, D. S., Ayyıldız, M. F. & Bakırdere, S. Liquid phase microextraction based sensitive analytical strategy for the determination of 22 hazardous aromatic amine products of azo dyes in wastewater and tap water samples by GC-MS system. Microchem J. 155, 104712 (2020).
Ràfols, C. & Barceló, D. Determination of mono- and disulphonated azo dyes by liquid chromatography–atmospheric pressure ionization mass spectrometry. J. Chromatogr. A. 777, 177–192 (1997).
Sun, H., Wang, F. & Ai, L. Determination of banned 10 azo-dyes in hot Chili products by gel permeation chromatography–liquid chromatography–electrospray ionization-tandem mass spectrometry. J. Chromatogr. A. 1164, 120–128 (2007).
Stauffer, E., Dolan, J. A. & Newman, R. Gas Chromatography and Gas Chromatography—Mass Spectrometry. in Fire Debris Anal. 235–293 (Elsevier, 2008). https://doi.org/10.1016/B978-012663971-1.50012-9
McEwen, C. N. & McKay, R. G. A combination atmospheric pressure LC/MS:GC/MS ion source: advantages of dual AP-LC/MS:GC/MS instrumentation. J. Am. Soc. Mass. Spectrom. 16, 1730–1738 (2005).
Boyd, J. M., Hrudey, S. E., Li, X. F. & Richardson, S. D. Solid-phase extraction and high-performance liquid chromatography mass spectrometry analysis of nitrosamines in treated drinking water and wastewater. TrAC Trends Anal. Chem. 30, 1410–1421 (2011).
Xiao, J. et al. Liquid-liquid extraction separation of lithium isotopes by using room-temperature ionic liquids-chloroform mixed solvent system contained benzo-15-crown-5. J. Mol. Liq. 223, 1032–1038 (2016).
Carabajal, M., Teglia, C. M., Cerutti, S., Culzoni, M. J. & Goicoechea, H. C. Applications of liquid-phase microextraction procedures to complex samples assisted by response surface methodology for optimization. Microchem J. 152, 104436 (2020).
Serbest, H., Bakırdere, S. & Keyf, S. Determination of silver in Metal Plating Wastewater by Slotted Quartz Tube Flame Atomic absorption spectrometry (SQT-FAAS) after preconcentration with Stearic Acid-Coated Magnetite nanoparticle-based solid-phase microextraction (SA-MNP-SPME). Anal. Lett. 55, 1104–1118 (2022).
Socas-Rodríguez, B., Herrera-Herrera, A. V., Asensio-Ramos, M. & Hernández-Borges, J. Dispersive Solid-Phase Extraction. in Analytical Separation Science 1525–1570 (Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, doi: (2015). https://doi.org/10.1002/9783527678129.assep056
Zhang, S., Wu, W. & Zheng, Q. Evaluation of modified Fe 3 O 4 magnetic nanoparticle graphene for dispersive solid-phase extraction to determine trace PAHs in seawater. Anal. Methods. 7, 9587–9595 (2015).
Zhang, X. et al. Graphene oxide-SiO 2 nanocomposite as the adsorbent for extraction and preconcentration of plant hormones for HPLC analysis. J. Chromatogr. B. 1046, 58–64 (2017).
Chen, X. & Ye, N. A graphene oxide surface–molecularly imprinted polymer as a dispersive solid-phase extraction adsorbent for the determination of cefadroxil in water samples. RSC Adv. 7, 34077–34085 (2017).
Khonkayan, K. et al. New approach for detection of chromate ion by preconcentration with mixed metal hydroxide coupled with fluorescence sensing of copper nanoclusters. Microchim Acta. 184, 2965–2974 (2017).
Peña-Méndez, E. M. et al. Metal organic framework composite, nano-Fe3O4@Fe-(benzene-1,3,5-tricarboxylic acid), for solid phase extraction of blood lipid regulators from water. Talanta 207, 120275 (2020).
Feng, X., Ding, X. & Jiang, D. Covalent organic frameworks. Chem. Soc. Rev. 41, 6010 (2012).
Yin, S. J. et al. Preparation of amino-functionalized covalent organic framework modified Fe3O4 nanoparticles for the selective enrichment of flavonoid glycosides. Microchem J. 164, 105990 (2021).
You, L. et al. Facile synthesis of Fe3O4@COF covalent organic frameworks for the adsorption of bisphenols from aqueous solution. J. Mol. Liq. 320, 114456 (2020).
Erarpat, S., Bodur, S. & Bakırdere, S. Nanoparticles Based Extraction Strategies for Accurate and Sensitive Determination of Different Pesticides. Critical Reviews in Analytical Chemistry at (2021). https://doi.org/10.1080/10408347.2021.1876552
de Dios, A. S. & Díaz-García, M. E. Multifunctional nanoparticles: Analytical prospects. Anal. Chim. Acta. 666, 1–22 (2010).
Deng, Z. H. et al. A core-shell structured magnetic covalent organic framework (type Fe3O4@COF) as a sorbent for solid-phase extraction of endocrine-disrupting phenols prior to their quantitation by HPLC. Microchim Acta. 186, 108 (2019).
Lin, G. et al. Room-temperature synthesis of core–shell structured magnetic covalent organic frameworks for efficient enrichment of peptides and simultaneous exclusion of proteins. Chem. Commun. 53, 3649–3652 (2017).
Li, W. T. et al. Fabrication of magnetic Fe3O4@metal organic framework@covalent organic framework composite and its selective separation of trace copper. Appl. Surf. Sci. 530, 147254 (2020).
Wang, H., Wang, R., Wang, L. & Tian, X. Preparation of multi-core/single-shell OA-Fe3O4/PANI bifunctional nanoparticles via miniemulsion polymerization. Colloids Surf. Physicochem Eng. Asp. 384, 624–629 (2011).
Zhao, Y., Zhu, Z., Liu, J., Liu, J. & Li, G. Magnetic solid-phase extraction followed by HPLC–DAD for highly sensitive determination of Phthalate Esters in Edible Vegetable oils. Food Anal. Methods. 14, 2375–2385 (2021).
Aghazadeh, M. et al. Cobalt hydroxide ultra-fine nanoparticles with excellent energy storage ability. Appl. Surf. Sci. 283, 871–875 (2013).
Chai, W., Wang, H., Zhang, Y. & Ding, G. Preparation of polydopamine-coated magnetic nanoparticles for dispersive solid-phase extraction of water-soluble synthetic colorants in beverage samples with HPLC analysis. Talanta 149, 13–20 (2016).
Sun, T., Sun, H. & Zhao, F. Dispersive solid-phase extraction for the determination of trace organochlorine pesticides in apple juices using reduced graphene oxide coated with ZnO nanocomposites as sorbent. J. Sep. Sci. 40, 3725–3733 (2017).
Duan, H. L. et al. Magnetically modified porous β-Cyclodextrin polymers for Dispersive solid-phase extraction high-performance liquid chromatography analysis of Sudan dyes. Food Anal. Methods. 12, 1429–1438 (2019).
Kartoğlu, B., Bahçivan, A., Erarpat, S., Bayraktar, A. & Bakirdere, S. Microwave assisted synthesis method for cobalt nanoleaves and its usage in sensitive determination of lead in blue butterfly tea extract and tap water samples by flame atomic absorption spectrophotometry. J. Food Compos. Anal. 121, 105373 (2023).
Magnusson, B. The fitness for purpose of analytical methods: a laboratory guide to method validation and related topics. Eurachem 57 (2014).
Tölgyesi, Á. & Sharma, V. K. Quantification of aromatic amines derived from azo colorants in textile by ion-pairing liquid chromatography tandem mass spectrometry. J. Chromatogr. B. 1137, 121957 (2020).
Sánchez, M. N., Santos, P. M., Sappó, C. P., Pavón, J. L. P. & Cordero, B. M. Microextraction by packed sorbent and salting-out-assisted liquid–liquid extraction for the determination of aromatic amines formed from azo dyes in textiles. Talanta 119, 375–384 (2014).
Cioni, F., Bartolucci, G., Pieraccini, G., Meloni, S. & Moneti, G. Development of a solid phase microextraction method for detection of the use of banned azo dyes in coloured textiles and leather. Rapid Commun. Mass. Spectrom. 13, 1833–1837 (1999).
Abdulra’uf, L. B., Hammed, W. A. & Tan, G. H. SPME fibers for the analysis of pesticide residues in fruits and vegetables: a review. Crit. Rev. Anal. Chem. 42, 152–161 (2012).
Kamenická, B. & Kuchtová, G. Critical review on electrooxidation and chemical reduction of azo dyes: economic approach. Chemosphere 363, 142799 (2024).
Vogl, J. Calibration strategies and Quality Assurance. in Inductively Coupled Plasma Mass Spectrometry Handbook 147–181 (Blackwell Publishing Ltd., Oxford, UK, https://doi.org/10.1002/9781444305463.ch4. (2009).
Anastas, P. T. & Warner, J. C. Principles of green chemistry. Green. Chem. Theory Pract. 29, 14821–14842 (1998).
Gałuszka, A., Migaszewski, Z. M., Konieczka, P. & Namieśnik, J. Analytical Eco-scale for assessing the greenness of analytical procedures. TrAC - Trends Anal. Chem. 37, 61–72 (2012).
Van Aken, K., Strekowski, L. & Patiny, L. EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters. Beilstein J. Org. Chem. 2, (2006).
Manousi, N., Wojnowski, W., Płotka-Wasylka, J. & Samanidou, V. Blue applicability grade index (BAGI) and software: a new tool for the evaluation of method practicality. Green. Chem. 25, 7598–7604 (2023).
Funding
No.
Author information
Authors and Affiliations
Contributions
Nazime Ebrar Karlıdağ: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. Sezin Erarpat Bodur: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft.Ömer Tahir Günkara: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. Sezgin Bakırdere: Conceptualization, Data curation, Investigation, Methodology, Supervision, Validation, Writing– review & editing.
Corresponding authors
Ethics declarations
Compliance with ethical standards
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Karlıdağ, N.E., Bodur, S.E., Günkara, Ö.T. et al. Simultaneous determination of nineteen azo dyes in water samples by gas chromatography-mass spectrometry after Fe3O4@COF based dispersive solid phase extraction. Sci Rep 14, 28406 (2024). https://doi.org/10.1038/s41598-024-80097-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-80097-8
