
Abstract
Taxanes, such as paclitaxel (PTX), stabilize microtubules and are used as a first-line therapy in multiple cancer types. Disruption of microtubule equilibrium, which plays an essential role in mitosis and cell homeostasis, ultimately results in cell death. Even though PTX is a very potent chemotherapy, its use is associated with major side effects and therapy resistance. Pelophen B (PPH), a synthetic analog of peloruside A, stabilizes microtubules through interaction with a non-taxoid binding site of β-tubulin. We evaluated the anticancer effect of PPH in a variety of tumor types by using established cell lines, early-passage cultures and ex vivo tumor-derived cultures that preserve the 3D architecture of the tumor microenvironment. PPH significantly blocks colony formation capacity, reduces viability and exerts additivity with PTX. Interestingly, PPH overcomes resistance to PTX. Mechanistically, PPH induces a G2/M cell cycle arrest and increases the presence of tubulin polymerization promoting protein (TPPP), inducing lysine 40 acetylation of α-tubulin. Although, results induced by paclitaxel or PPH are concordant, PPH’s unique microtubule binding mechanism enables PTX additivity and ensures overcoming PTX-induced resistance. In conclusion, PPH results in remarkable anti-cancer activity in a range of preclinical models supporting further clinical investigation of PPH as a therapeutic anticancer agent.
Similar content being viewed by others

Introduction
Taxanes are among the most commonly used chemotherapeutic drugs administered in neo-adjuvant, adjuvant and palliative setting. Paclitaxel (PTX) and docetaxel were introduced into clinical practice in the 90s and remain the cornerstone of treatment for multiple tumor types such as sarcoma, breast and gynecological malignancies1,2,3. Breast cancer is the most frequently diagnosed cancer in women and comprises 11.6% of all cancer cases4. On the other hand, the 5-year survival rate remains only 51% for ovarian cancer, even in well-developed countries such as the United States, and it is the sixth leading cause of cancer death among women5. Soft-tissue sarcomas are rare, heterogeneous cancers of which up to 80% of patients metastasize, with low response rates (15–20%) in metastatic setting6. Taxanes target microtubules, hollow cylindrical structures formed by the polymerization of a heterodimer of α- and β-tubulin. Taxanes bind to β-tubulin in the lumen of microtubules, leading to their stabilization (hence their name microtubule-stabilizing agents (MSA)). Given the importance of microtubule dynamics in mitosis and cell homeostasis, taxanes induce a G2/M cell cycle arrest, ultimately resulting in cell death. Unfortunately, regardless of the formulation type, approximately 60 to 70% of patients receiving taxanes experience peripheral neurotoxicity, significantly impacting health-related quality of life and long-term clinical effectiveness1. Clinical efficacy is further limited by the development of chemoresistance in patients receiving taxane treatment. Taxane chemoresistance can arise through several different mechanisms, including mutations at the taxoid binding site7. Taxane alternatives and/or adjusted formulations are under intense investigation8. Peloruside A (PLA), an MSA isolated from the marine sponge Mycale hentsheli, binds on the laulimalide/peloruside (LP) site located on the surface-exposed site of β-tubulin (Fig. 1a,b)9,10. Steinmetz and Prota performed an in-depth review summarizing the different MSA binding sites11,12. It has been shown that PLA’s mechanisms of action is very similar to PTX13,14,15 and PLA demonstrated potent antitumor activity both in in vitro as in vivo14,16,17,18,19. Additionally PLA synergizes with other MSAs20,21 and remains active in cell lines overexpressing the P-glycoprotein (Pgp) drug efflux pump or cells bearing mutations affecting the taxoid binding site18. Interestingly, PLA does not induce microtubule axial lattice expansion and likely does not disrupt recognition patterns for regulatory- and motor proteins implicated in peripheral neurotoxicity22. However, further clinical development of PLA is hindered by the scarcity of the natural product and the complexity and low product yield of its synthesis16.

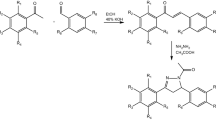
Binding site of PPH and chemical structure of PLA and PPH. (a) Microtubules are composed of α- and β-tubulin dimers. PTX binds on the luminal side of β-tubulin while PPH binds on the laulimalide/peloruside binding site on the microtubule surface. Lysine 40 acetylation (K40 Ac) of α-tubulin leads to stabilization of polymerized microtubules. Created with BioRender.com. (b) Chemical structure of PLA. (c) Chemical structure of PPH.
Pelophen B (PPH) (Fig. 1c), a structurally simplified analog of PLA, is a promising alternative to PLA, as its synthesis results in higher product yield as described in patent PCT/EP2014/075903. Similarly to PLA, PPH binds to the LP site suggesting PPH could overcome PTX chemoresistance (Fig. 1a). Moreover, PPH has better aqueous solubility than taxanes eliminating the need for a vehicle like Cremophor-EL, thus potentially reducing the side-effects such as hypersensitivity reactions22.
In this study, the anticancer activity of PPH is investigated in detail at the phenotypic and mechanistic level using established cell lines and early-passage or ex vivo tumor-derived cultures. In all experiments the activity of PPH is compared to PTX as this chemotherapeutic is widely used in clinical practice. Across both in vitro and ex vivo experiments, we demonstrate PPH effectively inhibits the proliferation of breast cancer, ovarian cancer and sarcoma cells. PPH also shows additivity upon combination with PTX. PPH hinders progression through the cell cycle by a G2/M arrest. Additionally, unbiased proteomic evaluation identified increased presence of tubulin polymerization-promoting protein (TPPP) responsible for lysine 40 (K40) acetylation of α-tubulin in PPH treated conditions. Finally, PPH overcomes PTX chemoresistance in vitro and ex vivo. In conclusion, our study warrants further research for the clinical development of PPH as a new therapeutic anticancer agent capable of overcoming PTX chemoresistance.
Results
PPH stabilizes microtubule polymerization
To ascertain the microtubule-stabilizing capacity of PPH, we conducted an in vitro microtubule polymerization assay. PTX, a well-known MSA, was used as positive control. Tubulin polymerization comprises three distinct stages: nucleation (lag phase), growth and steady state equilibrium. Equimolar doses of PPH or PTX increase the Vmax value of the growth phase by 2.92-fold and 3.08-fold, respectively, compared to the vehicle control. Both PPH and PTX block nucleation events. The polymer mass, measured by absorbance, reaches a slightly higher maximum for PPH with a maximal optical density (OD) of respectively 0.37 versus 0.33 for PTX. Under PPH treatment, the plateau level of polymerized tubulin mass is sustained for a prolonged time (Fig. 2a). From these findings we can conclude that PPH stabilizes microtubule polymerization with at least a similar efficacy as PTX.

PPH has antiproliferative activity in vitro and ex vivo. (a) Time-lapse tubulin polymerization assay showing PTX (5 µM) and PPH (5 µM) induce polymerization of microtubules in vitro and eliminate the nucleation phase. Data represent mean ± SD of three independent experiments. Statistical significance between groups was determined with one-way ANOVA and Tukey’s multiple comparisons test. (b) 3-day PPH treatment dose-dependently inhibits growth of cancer cell cultures with EC50 values in the low micromolar range indicated between brackets. (c) GRinf represents the normalized growth rate inhibition of the drug at infinite concentration and shows a cytotoxic effect (GRinf < 0), cytostatic (GRinf = 0) or partial growth inhibition (GRinf > 0) dependent on cell type. (d) Combining PTX and PPH leads to an additive effect in SKOV3 cells. Heatmap represents mean of 3 biological replicates (N = 3). δ-score < -10: antagonistic effect, -10 < δ-score < 10 additive effect, 10 < δ-score synergistic effect. Dotted square in 2-dimensional heatmap indicates the most synergistic area. (e) PPH reduces colony forming capacity of SKOV3 cells. Cells were treated with PTX (1 nM, 3 nM and 10 nM) or PPH (0.1 µM, 1 µM and 2.5 µM) . Samples were normalized to vehicle control. Bar plot represents mean ± SD of three biologically independent experiments. Statistical significance between groups was determined with one-way ANOVA and Tukey’s multiple comparisons test. (f) EC50 curve showing dose-dependent reduction in metabolic activity of SKOV3 spheroids after 5-day treatment with PTX or PPH. EC50 curve represents mean ± SD of two biologically independent experiments (N = 2) with each 3 technical replicates (n = 3). (g,h) Violin plot indicating the reduction in metabolic activity following 5 days of ex vivo treatment with PTX (0.1 µM) or PPH (50 µM) of 4T1 tumor fragments (g, n = 24) or soft tissue sarcoma (SAR183) (h, n = 16). Statistical significance between groups was determined with a Kruskal–Wallis test and Dunn’s multiple comparisons test. (i) Immunohistochemical Ki67 staining of ex vivo cultured 4T1 tumor fragments after 5 days of ex vivo treatment with PPH (50 µM). Images represent technical replicates. Levels of statistical significance are indicated as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P < 0.0001. DMSO was used as vehicle control.
PPH has in vitro antiproliferative activity
Next, the impact of PPH was dose-dependently investigated on a panel of established and early-passage patient-derived cell cultures from sarcoma, breast and ovarian cancer. The half maximum effective concentration (EC50) of PPH over all cell cultures tested reached the low µM range (mean 0.88 µM, 95% confidence interval: 0.55–1.21 µM) (Fig. 2b). Normalized growth rate inhibition of the drug at an infinite concentration (GRinf) indicated a high concordance between PTX and PPH with cytotoxic effects (GRinf < 0) in the majority of cell cultures (CAOV3, M28-2, MDA-MB-231, SAR030, SAR120 and SAR183) and partial growth inhibition (GRinf > 0) in 4T1 and caspase-3 deficient MCF7 cells (Fig. 2c). Next, cell viability was measured in a 10 × 10 dose–response matrix and SynergyFinder was used to detect possible synergism23. Since PPH and paclitaxel are both microtubule stabilizing agents, the Loewe synergy model was applied24,25. As expected from the complementary binding sites of PTX and PPH, combination treatment showed Synergy scores between -10 and 10, indicating additive effects in nine out of nine different cultures tested (Fig. 2d and Supplementary Fig. S1). However, specific dose combinations demonstrate synergy, e.g. SKOV3 cells treated with 1 µM PPH and 5 nM PTX showing a synergy score 16.8. Results based on metabolic ATP activity are further confirmed using assays measuring clonogenicity and three-dimensional (3D) growth of self-assembled cellular aggregates (spheroids), which better mimic tumor architecture due to the intrinsic oxygen and nutrient gradients. PPH dose-dependently blocks colony forming capacity of single-cell seeded cultures of SKOV3 and MCF7 (Fig. 2e and Supplementary Fig. S2). A metabolic ATP assay reveals dose-dependent viability reduction of SKOV3 spheroids with an EC50 value of 1 µM for PPH (Fig. 2f).
PPH has ex vivo antiproliferative activity
To bridge the translational gap between laboratory findings and clinical application, we tested ex vivo efficacy of PPH using tumor fragments from a 4T1 mammary cancer graft or a soft tissue sarcoma (SAR183) patient. Automated cutting of tumor tissue obtains size-controlled and -uniform rectangular hexahedral structures (length x width x height = 0.5 × 0.5 × 0.5mm = 0.125mm3) preserving the 3D architecture and spatial connection of the tumor microenvironment (TME), as such integrating more biology into therapeutic effectiveness26,27. Tumor fragments exhibit a significant reduction in viability after 5 days of ex vivo treatment with PTX or PPH (Fig. 2g,h). Immunohistochemical analysis at the endpoint reveals a decrease in Ki67-positive proliferating cancer cells suggesting drug efficacy in the presence of TME elements (Fig. 2i).
PPH induces G2/M cell cycle arrest
In asynchronous dividing adherent cell cultures of SKOV3, MCF7 and MDA-MB-231, EC50 concentrations of PPH maintain cell rounding following mitotic de-adhesion starting 6h after treatment (Supplementary Video S1-3). Time-dependent exposure with EC50 concentrations of PPH locks large proportions of the cell population in a rounded morphotype and to a higher extent than PTX at EC50 concentrations (Fig. 3a and Supplementary Fig. S3). The disturbed cell division by these rounded cells is confirmed through cell cycle analysis, revealing an accumulation in G2/M arrest in MCF7 cells which is more pronounced for PPH at 24h than PTX (Fig. 3b). This PPH-induced G2/M arrest is also observed in other cell cultures (Supplementary Fig. S3).

Mechanism of action of PPH. (a) PTX (3 nM) and PPH (1 µM) induce rounding of MCF7 breast cancer cells 24h post-treatment. White arrows indicate the rounded cells. Each biological replicate is represented with a different symbol (N = 3) and each symbol is a technical replicate (n = 3). Statistical significance between groups was determined with Kruskall Wallis test and Dunn’s multiple comparisons test. (b) 24-h treatment with PTX (100 nM) or PPH (5 µM) induces a G2/M cell cycle arrest in MCF7 breast cancer cells. Bar plot represents mean ± SD of three biologically independent experiments. Statistical significance between groups was determined with one-way ANOVA and Tukey’s multiple comparisons test. (c,d) Volcano plot depicting the differentially abundant proteins in SKOV3 spheroids after 5-day treatment with PPH (1 µM) (c) or PTX (4 nM) (d) compared to vehicle control. The vertical dotted red lines indicate a log2 fold change > 1. The horizontal dotted red line indicates a p-value = 0.05 (log10 adjusted p-value 1.30). Proteins are labelled with their corresponding gene names. Data are representative of the mean of six biological replicates (N = 6). (e) Graphs representing the upregulation of biological processes in vehicle treated spheroids versus PPH treated spheroids (top) or PPH treated spheroids versus vehicle treated spheroids (bottom) based on mass-spectometry assisted proteomics analysis and analysis with g:Profiler. (f,g) Heatmap of differentially regulated genes involved in cell cycling (GO: 0007049) (f) or microtubule-based processes (GO:0007017) (g) after PTX or PPH treatment versus vehicle control spheroids. (h) 24-h treatment with PTX (100 nM) or PPH (2.5 µm) of SKOV3 cells induces Lysine 40 acetylation (K40 Ac) of α-tubulin. GAPDH was used as loading control. Representative images from three biologically independent replicates are shown. Uncropped blots are shown in Supplementary Figure S5. Bar plot represents mean ± SD of three biologically independent experiments. Statistical significance between groups was determined with one-way ANOVA and Tukey’s multiple comparisons test. (i) Immunohistochemical K40 Ac of α-tubulin staining of ex vivo 4T1 tumor fragments after 5-day treatment with 100 nM PTX or 50 µM PPH. Levels of statistical significance are indicated as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P < 0.0001. DMSO was used as vehicle control.
PPH increases presence of TPPP and other proteins associated with microtubule stabilization
We use mass-spectrometry assisted proteomic analysis on spheroids to compare the effects between PPH and PTX at EC50 concentrations at the protein level. We decided to use 3D spheroids since they recapitulate tumor conditions more than 2D cell culture conditions. In accordance with previous results, vehicle-treated control spheroids express various proteins related to active cell cycling or normal microtubule dynamics (e.g. KIF14, KIF23, RACGAP1 and CENPC) and that while MSA treated spheroids contain a higher abundance of various isoforms of α- and β-tubulin, further confirming the microtubule-stabilizing capacity (Fig. 3c–g and Supplementary Fig. S4). Interestingly, TPPP is differentially abundant in PPH as well as PTX treated spheroids compared to control (Fig. 3c,d). TPPP promotes K40 acetylation of α-tubulin by binding to and inhibiting histone deacetylase 6 (HDAC6) and as such leads to more stable, polymerized microtubules and disturbed microtubule dynamics28. Indeed, Western blot analysis confirms a robust increase in the K40 acetylated α-tubulin protein levels following 24h treatment of ovarian cancer cells, especially after treatment with PPH (Fig. 3h). The increase in K40 acetylated α-tubulin protein was also demonstrated in ex vivo cultured 4T1 tumor fragments by endpoint immunohistochemical analysis after 5 days of ex vivo treatment with PTX and PPH (Fig. 3i).
PPH overcomes PTX resistance
A major problem in patients treated with PTX is acquired resistance. To investigate the impact of PPH on acquired PTX resistance we used a double strategy. First, we established SKOV3 ovarian cancer cells dose-dependently adapted to PTX and finally showing an resistance index (RI) of 18, indicating a 18-fold lower sensitivity (Fig. 4a). Despite this reduced taxane sensitivity, PPH potently decreases metabolic activity (EC50 = 3.2 µM, RI = 3.6) (Fig. 4b). PPH also decreases cell confluency (Fig. 4c,d) and colony formation capacity (Fig. 4e). Second, ex vivo cultured tumor fragments of a high-grade serous ovarian cancer (HGSOC), heavily pretreated with a combination of carboplatinum and paclitaxel, show higher sensitivity to PPH compared to PTX (Fig. 4f) and a significant increase in K40 acetylated α-tubulin (Fig. 4g). Both results using independent approaches indicate the capacity of PPH to overcome clinically relevant PTX resistance .

PPH overcomes PTX resistance. (a) PTX resistant phenotype of SKOV3 cells is confirmed by the increase of the EC50 value for PTX in SKOV3 PTX resistant cancer cells compared to parental SKOV3 cells. EC50 curve represents mean ± SD of three biologically independent experiments (N = 3) with each three technical replicates (n = 3). (b) PPH reduces viability of PTX resistant SKOV3 cells. EC50 curve represents mean ± SD of three biologically independent experiments (N = 3) with each three technical replicates (n = 3). (c,d) Real-time analysis of cell confluency of SKOV3 PTX resistant cells following PPH treatment. Microscopic pictures were taken every 6h and cell confluency was analyzed using IncuCyte software (indicated by yellow mask). Cell confluency curve represents mean ± SD of three biologically independent experiments (N = 3) with each 3 technical replicates (n = 3). Pictures are representative of three independent experiments. (e) Representative images and quantification of colony formation assay for SKOV3 PTX-resistant cells treated with PTX (1 nM, 3 nM and 10 nM) or PPH (0.1 µM, 1 µM and 2.5 µM) showing PPH overcomes PTX resistance. Samples were normalized to vehicle control (DMSO). Bar plot represents mean ± SD of three biologically independent experiments. Statistical significance between groups was determined with one-way ANOVA and Tukey’s multiple comparisons test. (f) Violin plot indicating the impact of 5-day treatment with PTX (0.1 µM) or PPH (50 µM) on ATP content of HGSOC ex vivo cultured tumor fragments. DMSO was used as vehicle control. Each symbol is a technical replicate (n = 22). Statistical significance between groups was determined with a Kruskal–Wallis test and Dunn’s multiple comparisons test. (g) Immunohistochemical K40 Ac of α-tubulin staining of ex vivo cultured patient-derived HGSOC tumor fragments after 5 days of PPH treatment (50 µM). Levels of statistical significance are indicated as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P < 0.0001. RI = resistance index. DMSO was used as vehicle control.
Discussion
Taxanes remain one of the most effective chemotherapeutic compounds for the treatment of various tumor types, such as sarcomas, breast and ovarian cancer. However, clinical efficacy is hindered by major side effects such as peripheral neuropathy and acquired chemoresistance. Research to find alternatives which are effective, safer and can overcome existing taxane chemoresistance, is an ongoing field of interest.
In this study we introduce PPH as a new chemotherapeutic with a similar activity and mode of action as PTX, but capable of overcoming PTX resistance, a clinically relevant problem resulting in poor treatment response and worse survival rates. PPH is a simplified fully synthetic molecule, compared to parent structurally complex PLA, in which the number of stereocenters is reduced from 10 to 6 and the tetrahydropyran ring is entirely replaced by an aromatic moiety22. Synthesis of PPH does not require the scarce natural product PLA, but is obtained via modular synthesis from commercial building block 3-bromofenyl acetonitrile, as described in patent PCT/EP2014/075903. We confirmed that PPH effectively stabilizes microtubules, to the same extent as the well-known MSA PTX. It should be noted that absorbance can be influenced by polymer quantity as well as polymer shape29. Consequently, future electron microscopy experiments should investigate whether PTX and PPH induce similarly shaped polymers, i.e. sheet polymers of tubulin versus microtubule polymers. The stabilization of microtubules can lead to different anti-proliferative properties. It was previously shown that PPH induces bundles of microtubules in interphase cells and abnormal multipolar mitotic spindles in metaphase cells15,22. Multipolar spindles, caused by MSAs, result in chromosome mis-segregation, activation of the spindle assembly checkpoint and eventually cell death30,31. Besides its role in mitotic cells, microtubule dynamics are also crucial for cell migration of, for instance, vascular endothelial cells. Therefore MSAs can inhibit angiogenesis aiding the elimination of the tumor and enhancing clinical effectiveness15,32. We demonstrate that PPH has clear antiproliferative capacity as in vitro experiments show a dose-dependent reduction in metabolic activity in various cell lines, covering sarcomas, breast and ovarian cancer. Since PPH binds to a different site on tubulin than the taxoid binding site, the possibility that PPH and PTX could synergize was tested9,10. Synergism or additivity could allow for lower treatment doses of each drug or shorter administration times, possibly resulting in less side effects. Combining PPH and PTX results in additive effects, with synergism only at specific dose combinations as a too low concentration would not elicit a response. In contrast, concentrations that are too high result in maximal effects before adding the second drug upon combination. These results are in line with expectations considering that these are both MSAs and that the structural congener PLA acts synergistically with PTX20,21. Additional follow-up studies should further explore optimal combination strategies, including a wider range of concentrations and different treatment schedules, and the pharmacodynamics of the combination. This would give valuable insights in maximization of therapeutic efficacy while minimizing side effects. The efficacy of PPH was further confirmed in 3D spheroids, since these simplified biomimetic models exhibit a diffusive gradient of nutrients and oxygen, causing metabolic gradients from the periphery to the inner core, resulting in spatial heterogeneity in proliferation, quiescence, necrosis and differentiation33,34. Another level of complexity was added by evaluating PPH in ex vivo cultured tissue fragments which preserve the TME and architecture. Voabil et al. demonstrated strong correlation between ex vivo and clinical responses to anti-PD1 treatment26. Notably, we show PPH remains effective and shows anti-tumor activity in both spheroid models as ex vivo tumor fragments of breast cancer and sarcoma making PPH a promising new anticancer therapeutic candidate.
To gain further insight into the mode of action, we performed flow cytometric analysis and show PPH induces a G2/M arrest, similarly to its structural congener PLA and PTX13,35. It has been long recognized that tubulin synthesis is self-regulated and that treatment with drugs that promote microtubule assembly increases the tubulin synthesis36,37,38. As expected, regardless of the type of MSA, mass-spectrometry-assisted proteomic analysis unveiled increased presence of various tubulin proteins. Proteomic analysis also revealed a higher abundance of TPPP in PPH and PTX treated cells. Interestingly, TPPP induces acetylation of α-tubulin by inhibiting histone deacetylase 6, ultimately stabilizing microtubules28,39. Accordingly, we find increased protein levels of K40 acetylated α-tubulin protein after PPH treatment, both in vitro as ex vivo. It should be considered that the increase in K40 acetylated α-tubulin could, as a result from the self-regulation of tubulin synthesis, be partly caused by an increase in total α-tubulin. Nonetheless it is an interesting observation and future studies should elicit the relevance of K40 acetylation of α-tubulin in the mechanism of action of PPH and PTX. This could be achieved by combining PPH with selective HDAC6 inhibitors such as Tubathian A40. In doing so the effect of K40 acetylation on microtubule dynamics or cancer cell behavior such as proliferation, migratory or invasive activity, can be further explored. Overall, we demonstrate that PTX and PPH elicit very similar effects across all performed assays but attention must be drawn to the fact that PPH, except for the polymerization assay, is approximately 100-fold less potent than PTX. With the current knowledge available we can only speculate about the potential causes but they may include differences in plasma membrane penetration, chemical stability and structural modifications of microtubules during the microtubule stabilization phase.
Up to 50% of breast cancer patients and almost all recurrent ovarian cancer patients develop taxane resistance, impeding therapeutic efficacy41. We demonstrate that PPH effectively reduces metabolic activity and cell confluency in PTX resistant ovarian cancer cells. This is in line with a study from Estévez-Gallego et al. where PPH remained active in A2780AD cells, an ovarian cancer multidrug resistant cell line overexpressing the Pgp drug transporter. A2780AD showed resistance to PTX (IC50 = 1.6 µM, RI 1100) while retaining PPH activity (IC50 = 7.4 µM, RI index 5.2)22. Moreover, this in conjunction with a study from Gaitanos et al. demonstrating that PLA remained active in Chinese hamster ovary (CHO) cells overexpressing the Pgp drug transporter as well as A2780AD cells18. Although not investigated in this study, we hypothesize that PPH, similar to PLA18, will remain active in the presence of mutations affecting the taxoid binding site of β-tubulin. Additionally, we demonstrate that PPH remains active in ex vivo cultured tumor HGSOC tumor fragments from a patient heavily pretreated with carboplatinum and paclitaxel.
Besides taxane resistance, clinical effectiveness of taxanes is hindered by taxane-induced peripheral neuropathy (TIPN). Preclinical evidence indicates that TIPN is linked to structural modifications on the microtubule filament upon taxane binding42,43. These modifications could disrupt the recognition patterns for regulatory microtubule-associated proteins (MAPs) and motor proteins, perturbing the neural function44. However, it was recently discovered that PPH does not induce axial lattice expansion of microtubules, indicating that PPH does not significantly affect microtubule structure22. This implicates not only that PPH antitumor treatment potentially results in fewer neurotoxicity compared to PTX but also could be a promising therapeutic strategy for neurodegenerative diseases caused by compromised microtubule structures, such as Alzheimer’s disease45,46.
We provide compelling evidence for the anticancer potential of PPH. However, our study has some limitations. Our study focuses on and demonstrates preclinical effectiveness of PPH, but further in vivo studies are needed to assess in vivo safety and efficacy. Additionally, the underlying mechanism of overcoming PTX resistance is not fully elucidated and further research through transcriptomic or proteomic analyses is required to identify the involved pathways.
In conclusion, exploitation of microtubule stabilization by PPH, through binding the surface-exposed site of β-tubulin, results in remarkable anti-cancer activity in a range of preclinical cell culture models. Additionally, PPH demonstrates activity in ex vivo tumor fragments and retains sensitivity in PTX-resistant in vitro as well as ex vivo models. While more extensive research is needed to assess in vivo safety, (neuro)toxicity and efficacy, our findings support further clinical development of PPH as a therapeutic anticancer agent.
Methods
Cell culture
The chondrosarcoma cell line SW-1353 (cat. no. HTB-94), high-grade ovarian carcinoma cell lines CAOV3 (cat. no. HTB-75) and SKOV3 (cat. no. HTB-77), caspase-3 deficient breast cancer MCF7 cells (cat. no. HTB-22), estrogen receptor (ER) negative breast cancer cell lines MDA-MB-231 (cat. no. HTB-26) and 4T1 (cat. no. CRL-2359) were obtained from the American Type Culture Collection (ATCC). Early passage cell cultures SAR030 (undifferentiated pleiomorphoc sarcoma) and SAR183 (malignant peripheral nerve sheet sarcoma, location A4) were derived from patient sarcoma samples47,48. The early passage low-grade serous ovarian carcinoma patient-derived cell line M28-2 was isolated as described by De Thaye et al.49 All cell lines, except for M28-2 were routinely cultured in complete DMEM high glucose (DMEM-HG, 25 mM glucose, cat. no. 41965039, ThermoFisher) supplemented with 10% heat-inactivated FBS (Pan Biotech, cat. no. P30-3306), 100 IU/ml penicillin and 100 µg/ml streptomycin (cat. no. 15070063, ThermoFisher). M28-2 was cultured in EMEM (cat. no. 10–009-CV, Corning) supplemented with 10% heat-inactivated FBS (Pan Biotech, cat. no. P30-3306), 100 IU/ml penicillin and 100 µg/ml streptomycin (cat. no. 15070063, ThermoFisher). Cell cultures were monthly tested with Mycoalert Mycoplasm Detection Kit (cat. no. LT07-318, Lonza). All cell lines were maintained as monolayers at 37°C in a humidified incubator, in an atmosphere of 5% CO2 in air and passaged at 80% confluence. During experiments cell lines were cultured in DMEM low glucose (DMEM-LG, 5.6 mM glucose, cat. no. 11564446, ThermoFisher) supplemented with 10% heat-inactivated FBS (Pan Biotech, cat. no. P30-3306), 100 IU/ml penicillin and 100 µg/ml streptomycin (ThermoFisher, cat. no. 15070063).
Compounds
Drugs used were paclitaxel (PTX) (cat.no. HY-B0015_10mM, Bioconnect) and Pelophen B (PPH) (synthesized by the Department of Organic and Macromolecular Chemistry at Ghent University). All compounds were dissolved in dimethyl sulfoxide (DMSO). Stock solution of PTX was stored at − 80°C. PPH stock solutions were stored at − 20°C. Drugs containing media at given concentrations were made fresh for each experiment. DMSO was used as a vehicle control where appropriate.
Generation of paclitaxel resistant cell line
The PTX resistant SKOV3 cell line was derived from the parental SKOV3 cell line. Parental SKOV3 cells were exposed to a low starting concentration of 0.1 nM PTX and the concentration was increased by 0.1 nM at weekly intervals, until a dose of 1 nM was reached. Thereafter the dose was weekly increased by 1 nM increments until a final dose of 3 nM was reached. To maintain the resistant phenotype the cells were, in routine cultures, continuously exposed to 3 nM PTX.
Microtubule polymerization assay
Polymerization of porcine tubulin was monitored using a tubulin polymerization assay kit (cat.no. BK006P, Cytoskeleton Inc) according to manufacturers’ instructions. Briefly, purified tubulin (3 mg/ml) in a buffer (80 mM PIPES, 2mM MgCl2, 0.5 mM EGTA, 15% glycerol and 1mM GTP) was incubated at 37°C in the presence of either vehicle (DMSO), 5 µM PTX or 5µM PPH. Tubulin polymerization was monitored by absorbance readout (HTX Synergy, BioTek and Gen5 Data Analysis Software v3.08.01) for 60 min at 37°C and 340 nm every 60s. The data represented is the mean of 3 independent experiments, error bars indicate standard deviation (SD). Statistical significance of AUC was determined with a one-way ANOVA and Tukey’s multiple comparison test with an alpha level of significance set at 0.05.
ATP assay with CellTiter Glo 2.0
Drug sensitivity in monolayer cultured cells was determined by the CellTiter-Glo 2.0 metabolic activity assay (cat.no. G9243, Promega). Cells were seeded in 384-well plates (cat.no. CLS3765, Corning). After overnight attachment cells were treated with PTX and/or PPH for 3 days at 37°C (5% CO2 in normoxia). Then Cell Titer Glo 2.0 reagent was added according to manufacturer’s instructions. The contents were, shielded from light, mixed on an orbital shaker to induce cell lysis. After 20 min of incubation (room temperature), luminescence readout was performed using a spectrophotometer microplate reader (BioTek) using Gen5 Data Analysis Software (v3.08.01) at 590 nm. All data are the means ± SD of three biological replicates (N = 3) per condition, with three technical replicates (n = 3) for each biological replicate. Viability is reported relative to vehicle control (DMSO). EC50 values were determined in Graphpad Prism (v8.4.3). Growth rate inhibition metrics (GRinf) were calculated using GRcalculator50. Synergy analysis was performed using SynergyFinder23. Cell morphology analysis was performed using ImageJ software (version 1.54f.).
Cell cycle analysis
SKOV3 and MCF7 cells were treated with 0.05% DMSO, 100 nM PTX or 5 µM PPH for 24h prior to fixation with ice-cold ethanol. Fixed cells were resuspended in propidium iodide solution (PI, 50 µg/ml) (cat. no. P4864, Sigma Aldrich, supplemented with RNAse (100 µg/ml) (cat. no. 10109142001, Roche). Thereafter, cellular DNA content was determined using flow cytometry (FACS Symphony, BD). Cell cycle distribution was analyzed using FlowJo v10.9 software. Analysis was performed on three biological replicates (N = 3) per condition. Statistical significance between groups was determined with a one-way ANOVA and Tukey’s multiple comparison test or unpaired two-tailed t-test with an alpha level of significance set at 0.05 .
Colony formation assay
To examine colony forming ability cells were seeded in 6-well plates in 3 ml DMEM LG (5.6 mM glucose, cat. no. 11564446, ThermoFisher) supplemented with 10% heat-inactivated FBS (Pan Biotech, cat. no. P30-3306), 100 IU/ml penicillin and 100 µg/ml streptomycin (ThermoFisher, cat. no. 15070063). After overnight attachment, drugs were added to each well (0.1% DMSO, 1 nM PTX, 3 nM PTX, 10 nM PTX, 0.1 µM PPH, 1 µM PPH or 2.5 µM PPH) and the plates were incubated at 37°C (5% CO2 and normoxia). Following three days of incubation in the presence of drugs, cells from each treatment condition were split, reseeded at a density of 250 cells in 3 ml complete drug-free culture medium and allowed to form colonies during 10 days to assess colony forming capacity. Crystal violet staining was used as an endpoint measurement. The experiment was carried out in three biological replicates (N = 3), error bars indicate SD. Statistical analysis between groups was determined with a one-way ANOVA and Tukey’s multiple comparison test with an alpha level of significance set at 0.05.
Western blot
SKOV3 cells were treated with PTX (final concentration 100 nM) and PPH (final concentration 2.5 µM) for 24 h. DMSO was used as vehicle control. Control and treated cells were collected in 1.5 laemmli buffer (aquadest containing 0.125M Tris–HCl, 15% glycerol and 3.45% SDS, pH 6.8) and ultrasonicated (3 times 10 s, amplitude 30%). After determining protein concentrations with DC protein assay (cat. no. 5000113, 5,000,114, 5,000,115 , Bio-Rad) and BioDrop (Harvard Bioscience Inc.), cell lysates were suspended in reducing sample buffer (aquadest containing 0.125M Tris–HCl, 15% glycerol, 3.45% SDS, 5% β-mercaptoethanol, 5% bromophenol blue 0.25%, pH 6.8) and boiled for 5 min at 95°C. Protein lysates (20 µg) were separated on a 12% SDS-PAGE gel and transferred to nitrocellulose membrane (cat. no. 162–0115, Bio-Rad). Non-specific binding sites were blocked using 5% non-fat milk in phosphate buffered saline (PBS) with 0.5% Tween-20 (cat. no. P1379, Merck Life Science). To evaluate K40 acetylated α-tubulin expression membranes were incubated overnight at 4°C with anti-acetylated K40 α-tubulin 1:1000 (cat. no. ab179484, Abcam) or anti α-tubulin 1:1000 (cat.no. T5168, Merck Life Science), diluted in 5% non-fat milk in PBS supplemented with 5% Tween-20 (cat. no. P1379, Merck Life Science). GAPDH (1:2000, cat. no. G8795, Merck Life Science) was used as loading control. After washing the membranes, incubation with anti-mouse IgG (1:3000, cat. no. NA931, Cytiva) or anti-rabbit IgG (1:3000, cat. no. NA934, Cytiva) was performed at room temperature for 1 h. Clarity Max Western ECL kit (cat. no. 1705062, Bio-Rad) was used following manufacturer’s instructions to capture the luminescent signal using the Invitrogen iBright Imaging System (Thermo Fisher Scientific). Uncropped blots can be found as in supplementary information (Supplementary Fig. S5).
SKOV3 spheroid formation and drug treatment
Cells were seeded in 384-well ULA plates (cat. no. MS-9384UZ, S-bio) in a seeding density of 2000 SKOV3 cells in 40 µl cell culture media per well and allowed to form spheroids during 48h (37°C, 5% CO2, normoxia). Subsequently spheroids were supplemented with 40 µl cell culture medium containing PTX or PPH. To prevent evaporation 384-well plates were sealed with Breathe-Easy semipermeable membranes. Cells were cultivated for 5 more days (37°C, 5% CO2) under normoxic conditions.
Ex vivo tumor fragments
High grade serous ovarian carcinoma (HGSOC) tumor fragments were obtained from the patient at the time of surgery, with informed written patient consent in accordance with the regulatory guidelines of the Ethics Committee of Ghent University Hospital. A pathologist macroscopically selected solid tumor lesions from the resected tumor material and placed the material in ice-cold DMEM LG (cat. no. 31885023, ThermoFisher ) supplemented with 10% FBS (Sigma Aldrich), 100 U/ml penicillin and 100 µg/ml streptomycin (cat. no. 15070063, Life Technologies). All animal experiments were conducted in accordance with the regulatory guidelines of the Ethics Committee of Ghent University Hospital and the ARRIVE guidelines. All animals were purchased from Charles River Laboratories. All animals were housed in a ventilated isolation environment with a 12-h light/dark cycle, 40% to 70% humidity, a temperature range of 21 to 25°C and ad libithum access to food and water. Patient-derived SAR183 xenografts were established by implantation of tumor tissue of patient sample SAR183, location A4. Implantation was performed orthotopically in a female NOD scid gamma (NSG) mouse (6–8 weeks old, Charles River laboratories) and the tumor was resected after the primary tumor reached a volume of 250-500mm3. This first-generation patient-derived xenograft tissue was then orthotopically implanted in a new female NSG mouse (n = 1) to create a second generation and was resected for experimental use after a volume of 500mm3 was reached. The animal was euthanized using cervical dislocation under isoflurane (inhalation) anesthesia. For the 4T1 xenograft a female BALB/cBYJ mouse (n = 1), 5 to 6 weeks old was orthotopically injected with 5 × 105 4T1 cells (1:1 serum free DMEM medium: Matrigel (cat.no. 354234, Corning)) in the left mammary fat pad. Once the tumor volume reached 1000 mm3, the mouse was euthanized by cervical dislocation under isoflurane (inhalation) anesthesia, and the 4T1 xenograft tumor was resected for experimental use. All tumor material was immediately processed by automated cutting into small tumor fragments of 0,125 mm3 and placed on ice. After processing the tumor fragments from different regions within a tumor were mixed to ensure uniform representation of the tumor. Thereafter individual tumor fragments were seeded in pre-warmed (37°C) DMEM LG (cat. no. 31885023, ThermoFisher ) supplemented with 10% FBS (Sigma Aldrich), 100 U/ml penicillin (Life Technologies) and 100 µg/ml streptomycin (Life Technologies) and treated with PTX or PPH with a final concentration of 100 nM and 50 µM respectively. Control cultures were treated with 0.1% DMSO. To prevent evaporation 96-well (cat. no. MS-9096UZ, S-bio) plates were sealed with Breathe-Easy semipermeable membranes (cat. no. Z380059, Merck Life Science). Tumor fragments were cultivated up to 5 days (37°C, 5% CO2) under normoxic conditions.
ATP assay with CellTiter-Glo 3D
Drug sensitivity in spheroids and ex vivo tumor fragments was determined by the CellTiter-Glo 3D metabolic activity assay (cat.no. G9683, Promega). Every spheroid or tumor fragment was transferred to a white, flat-bottom 96-well plate (cat. no. 10479501, Fisher Scientific). An equal amount of CellTiter Glo 3D reagent was added to the wells according to manufacturer’s instructions. The contents were, shielded from light, mixed on an orbital shaker for 5 min to induce cell lysis and thereafter incubated during 25 min (room temperature). For better penetration of tumor fragments the CellTiter Glo 3D reagent was supplemented with 1% Triton X-100. Luminescence readout was performed using a spectrophotometer microplate reader (HTX Synergy, BioTek and Gen5 Data Analysis Software v3.08.01) at 590 nm. Data are the mean ± SD. Viability is reported relative to vehicle control (DMSO). EC50 values of both drugs were determined in Graphpad Prism.
Proteomics
Following treatment SKOV3 spheroids were resuspended in lysis buffer comprising of 50 mM TEAB (cat. no. T7408, Merck) and 5% SDS (cat. no. 71725, Merck) in Pierce Water (cat. no. 51140, Fisher Scientific). Then spheroids were ultrasonicated (3 times 10 s, amplitude 30%) and centrifuged. Supernatant containing the proteins was collected and stored at -20°C upon further processing. After thawing, protein concentrations were measured with a plate reader (HTX Synergy, BioTek and Gen5 Data Analysis Software v3.08.01) using the Pierce 600nm protein assay (cat. no. 22662, Life Technologies). Each sample was reduced with 200 mM DTT (cat. no. 20290, Thermo Scientific), then the solution was incubated first at 95 °C for 10 min and then at 60 °C for 1 h while shaking. The reduced proteins were alkylated with 200 mM iodoacetamide (IAA) (cat. no. I1149, Merck) in the dark for 30 min. Then samples were acidified with 12% aqueous phosphoric acid (cat. no. 345245, Merck) and mixed with 6 × volumes of 90% methanol (cat.no. 1.06035.1000, Merck), 100 mM TEAB, pH 7.1 (S-Trap binding buffer). The samples were loaded onto the filter of the S-Trap micro column (C02-micro-80, ProtiFi) and centrifuged at 4000rpm for 30 s. The flowthrough was discarded each time and the samples were washed 9 times with the S-Trap binding buffer. Proteins trapped in the S-Trap column were digested with Trypsin/Lys-C Mix (cat. no. V5073, Promega) at 47°C for 1 h at a protein-to-enzyme ratio of 1:25 (w/w). The digested peptides were eluted with buffers in order: 50 mM TEAB pH 8.0, 0.2% aqueous formic acid (cat. no. 28905, Thermo scientific), and 50% acetonitrile (cat.no. 34967, Fluka)/0.2% aqueous formic acid. Then the samples were lyophilized using a SpeedVac (Thermo Fisher Scientific) with cold trap and desalted with C18 tips (cat. no. 66883, Empore). Finally, the desalted peptides were again SpeedVac dried and dissolved in 0.1% formic acid for liquid chromatography-mass spectrometry analysis. Equal peptide concentrations were analyzed by a nanoflow HPLC system (Easy-nLC1200, Thermo Fisher Scientific) coupled to Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a high-field asymmetric waveform ion mobility spectrometry (FAIMS) device between a nano-electrospray ionization source and the mass spectrometer. Compensation voltages of -50 V and -70 V were used. Peptides were first loaded on a trapping column and then separated inline on a 15 cm C18 column (75 μm × 15 cm, ReproSil-Pur 3 μm 120 Å C18-AQ, Dr. Maisch HPLC GmbH, Ammerbuch-Entringen, Germany) with a 120-min 2-step gradient consisting of solvents A (0.1% formic acid) and solvent B (acetonitrile/water (95:5(v/v)) with 0.1% formic acid) from 6–21% B in 62 min, 21–36% B in 48 min, 36–100% in 5 min and held at 100% B for 5 min. All samples were analyzed by data independent acquisition (DIA), in which each cycle consisted of a full scan of 345–1005 m/z at a resolution of 120,000, maximum injection time of 50 ms and automatic gain control (AGC) target of 7e5. DIA MS2 scans were employed for all DIA scans at a resolution of 30,000, AGC target 1e6, maximum injection time of 52 ms and 30 windows of variable window scheme with an overlap of 1 m/z covering m/z range from 350 to 1000. DIA spectra were acquired at both -50 V and -70 V FAIMS compensation voltages. Identified proteins were analyzed and visualized using R (version 4.2.3). Pathway enrichment analysis(gene ontology) was performed using g:Profiler51.
IHC
Immunohistochemistry was performed on 5 µm sections from formalin-fixed paraffin-embedded tumor fragments of 4T1 xenograft and HGSOC. Deparaffinization and antigen activation of the sections were achieved by heat treatment. The IHC process was conducted using the ultraView Universal DAB detection kit (cat. no. 760–500, Ventana), an automated staining instrument (BenchMark ULTRA IHC/ISH System, Roche), and primary monoclonal antibodies against acetyl-alpha tubulin (1:2000, cat. MABT868, clone 6-11B1, Sigma Aldrich) and Ki67 (ready-to-use, cat. 790–4286, clone 30–9, Roche).
Data analysis
All data are presented as mean ± SD. Statistical analysis was conducted using Graphpad Prism 8 software (v8.4.3). Outliers were calculated using the ROUT method (Q = 1%). Shapiro–Wilk assessment for normality was conducted and unpaired comparisons using a Kruskal–Wallis or one-way ANOVA were performed with Dunn’s or Tukey’s multiple comparisons test. P value < 0.05 was considered statistically relevant. Levels of statistical significance are indicated as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P < 0.0001. Detailed p-values can be found in supplementary materials.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD049685. There are restrictions to the availability of Pelophen B (PPH) due to patent PCT/EP2014/075903. All other data presented in the study are available on request from the corresponding author.
References
Jordan, M. A. & Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265 (2004).
Yang, C. P. H. & Horwitz, S. B. Taxol®: The first microtubule stabilizing agent. Int. J. Mol. Sci. 18, 1733 (2017).
Tian, Z. & Yao, W. Albumin-bound paclitaxel: Worthy of further study in sarcomas. Front. Oncol. 12, 815900 (2022).
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49 (2024).
Gounder, M. M. et al. Clinical genomic profiling in the management of patients with soft tissue and bone sarcoma. Nat. Commun. 13, 3406 (2022).
Rodrigues-Ferreira, S., Moindjie, H., Haykal, M. M. & Nahmias, C. Predicting and overcoming taxane chemoresistance. Trends Mol. Med. 27, 138–151 (2021).
Mosca, L., Ilari, A., Fazi, F., Assaraf, Y. G. & Colotti, G. Taxanes in cancer treatment: activity, chemoresistance and its overcoming. Drug Resist. Updat. 54, 100742 (2021).
Huzil, J. T. et al. A unique mode of microtubule stabilization induced by Peloruside A. J. Mol. Biol. 378, 1016–1030 (2008).
Prota, A. E. et al. Structural basis of microtubule stabilization by Laulimalide and Peloruside A. Angew. Chem. Int. Ed. Engl. 53, 1621–1625 (2014).
Steinmetz, M. O. & Prota, A. E. Microtubule-targeting agents: strategies to hijack the cytoskeleton. Trends Cell Biol. 28, 776–792 (2018).
Steinmetz, M. O. & Prota, A. E. Structure-based discovery and rational design of microtubule-targeting agents. Curr. Opin. Struct. Biol. 87, 102845 (2024).
Hood, K. A. et al. Peloruside A, a novel antimitotic agent with paclitaxel-like microtubule-stabilizing activity. Cancer Res. 62, 3356–3360 (2002).
Chan, A., Andreae, P. M., Northcote, P. T. & Miller, J. H. Peloruside A inhibits microtubule dynamics in a breast cancer cell line MCF7. Invest. New Drugs 29, 615–626 (2011).
Ganguly, A., Cabral, F., Yang, H. & Patel, K. D. Peloruside A is a microtubule-stabilizing agent with exceptional anti-migratory properties in human endothelial cells. Oncoscience 2, 585–595 (2015).
Meyer, C. J. et al. Peloruside A inhibits growth of human lung and breast tumor xenografts in an athymic nu/nu mouse model. Mol. Cancer Ther. 14, 1816–1823 (2015).
Kanakkanthara, A. et al. βI-tubulin mutations in the laulimalide/peloruside binding site mediate drug sensitivity by altering drug-tubulin interactions and microtubule stability. Cancer Lett. 365, 251–260 (2015).
Gaitanos, T. N. et al. Peloruside A does not bind to the taxoid site on bèta-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res. 64, 5063–5067 (2004).
Řehulka, J. et al. Peloruside A-induced cell death in hypoxia Is p53 dependent in HCT116 colorectal cancer cells. J. Nat. Prod. 81, 634–640 (2018).
Wilmes, A., Bargh, K., Kelly, C., Northcote, P. T. & Miller, J. H. Peloruside A synergizes with other microtubule stabilizing agents in cultured cancer cell lines. Mol. Pharm. 4, 269–280 (2006).
Wilmes, A. et al. Synergistic interactions between peloruside A and other microtubule-stabilizing and destabilizing agents in cultured human ovarian carcinoma cells and murine T cells. Cancer Chemother. Pharmacol. 68, 117–126 (2011).
Estévez-Gallego, J. et al. Chemical modulation of microtubule structure through the laulimalide/peloruside site. Structure 31, 88–99 (2023).
Ianevski, A., Giri, A. K. & Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 50, W739–W743 (2022).
Yadav, B., Wennerberg, K., Aittokallio, T. & Tang, J. Searching for drug synergy in complex dose–response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 13, 504–513 (2015).
Loewe, S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 3, 285–290 (1953).
Voabil, P. et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med. 27, 1250–1261 (2021).
Van Renterghem, A. W. J., Van De Haar, J. & Voest, E. E. Functional precision oncology using patient-derived assays: Bridging genotype and phenotype. Nat. Rev. Clin. Oncol. 20, 305–317 (2023).
Tőkési, N. et al. TPPP/p25 promotes tubulin acetylation by inhibiting histone deacetylase 6. J. Biol. Chem. 285, 17896–17906 (2010).
Mirigian, M., Mukherjee, K., Bane, S. L. & Sackett, D. L. Measurement of In vitro microtubule polymerization by turbidity and fluorescence. in Methods in Cell Biology vol. 115 215–229 (Academic Press Inc., 2013).
Zasadil, L. M. et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 6, 229ra43 (2014).
Bekier, M. E., Fischbach, R., Lee, J. & Taylor, W. R. Length of mitotic arrest induced by microtubule-stabilizing drugs determines cell death after mitotic exit. Mol. Cancer Ther. 8, 1646–1654 (2009).
Bocci, G., Di Paolo, A. & Danesi, R. The pharmacological bases of the antiangiogenic activity of paclitaxel. Angiogenesis 16, 481–492 (2013).
Hirschhaeuser, F. et al. Multicellular tumor spheroids: an underestimated tool is catching up again. J. Biotechnol. 148, 3–15 (2010).
Riedl, A. et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J. Cell Sci. 130, 203–2018 (2017).
Pera, B. et al. Molecular recognition of peloruside A by microtubules. The C24 primary alcohol is essential for biological activity. Chembiochem. 11, 1669–1678 (2010).
Ben-Ze’ev, A., Farmer, S. R. & Penman, S. Mechanisms of regulating tubulin synthesis in cultured mammalian cells. Cell 17, 319–325 (1979).
Cleveland, D. W., Lopata, M. A., Sherline, P. & Kirschner, M. W. Unpolymerized tubulin modulates the level of tubulin mRNAs. Cell 25, 537–546 (1981).
Theodorakis, N. G. & Cleveland, D. W. Physical evidence for cotranslational regulation of β-Tubulin mRNA degradation. Mol. Cell Biol. 12, 791–799 (1992).
Al-Bassam, J. & Corbett, K. D. α-Tubulin acetylation from the inside out. Proc. Natl. Acad. Sci. USA 109, 19515–19516 (2012).
Depetter, Y. et al. Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models. Int. J. Cancer 145, 735–747 (2019).
Maloney, S. M., Hoover, C. A., Morejon-Lasso, L. V. & Prosperi, J. R. Mechanisms of taxane resistance. Cancers (Basel) 12, 3323 (2020).
Kellogg, E. H. et al. Insights into the distinct mechanisms of action of taxane and non-taxane microtubule stabilizers from cryo-EM structures. J. Mol. Biol. 429, 633–646 (2017).
Manka, S. W. & Moores, C. A. The role of tubulin–tubulin lattice contacts in the mechanism of microtubule dynamic instability. Nat. Struct. Mol. Biol. 25, 607–615 (2018).
Gornstein, E. & Schwarz, T. L. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology 76, 175–183 (2014).
Brunden, K. R., Lee, V. M. Y., Smith, A. B., Trojanowski, J. Q. & Ballatore, C. Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol. Dis. 105, 328–335 (2017).
Ballatore, C., Lee, V. M. Y. & Trojanowski, J. Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 (2007).
Peirsman, A. et al. MISpheroID: a knowledgebase and transparency tool for minimum information in spheroid identity. Nat. Methods 18, 1294–1303 (2021).
Fischer, S. et al. Post-operative minimal residual disease models to study metastatic relapse in soft-tissue sarcoma patient-derived xenografts. Clin. Transl. Med. 13, e1290 (2023).
De Thaye, E. et al. Establishment and characterization of a cell line and patient-derived xenograft (PDX) from peritoneal metastasis of low-grade serous ovarian carcinoma. Sci. Rep. 10, 6688 (2020).
Hafner, M., Niepel, M., Chung, M. & Sorger, P. K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 13, 521–527 (2016).
Kolberg, L. et al. g:Profiler-inter operable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 51, W207–W212 (2023).
Acknowledgements
This work was supported from the Concerted Research Actions and Industrial Research Fund from Ghent University.
Author information
Authors and Affiliations
Contributions
SV and ODW designed the study and interpreted the data. OR and JVdE designed and synthesized Pelophen B. SV and EB performed the experiments. SV analyzed the data. ZX provided murine tissue. SE, SG, GS, PT, WC, HaDe and HeDe provided human tissue for ex vivo experiments. SV, EB, PR, JH, SD and ODW performed, analyzed and interpreted the proteomics experiments. LC and PD performed the immunohistochemical stainings. AH and ODW supervised all experiments. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
Johan Vander Eycken is patentholder of Pelophen B (PCT/EP2014/075903). All other authors declare no competing interests.
Ethics approval and consent to participate
Informed consent of patients to use tumor material was obtained after the study protocol was approved by the institutional review board of the Ghent University Hospital (EC 2018/0080 and ONZ-2022–0345, EC 2019/1869 and ONZ-2023-0502). All animal experiments were approved by the local ethics committee (ECD 19-45 and ECD 19-86, Ghent University Hospital). All animal experiments were conducted in accordance with the regulatory guidelines of the Ethics Committee of Ghent University Hospital and the ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vermeulen, S., Ernst, S., Blondeel, E. et al. Pelophen B is a non-taxoid binding microtubule-stabilizing agent with promising preclinical anticancer properties. Sci Rep 14, 30188 (2024). https://doi.org/10.1038/s41598-024-80672-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-80672-z
Keywords
This article is cited by
-
Development of the fluorescent probe CenSpark for labeling centrioles and cilia
Nature Chemical Biology (2026)
