
Abstract
The early microbial colonization of the porcine gut is an important priming factor for gut and immune development. Nevertheless, little is known about the composition of microbes that translocate into the ileo-cecal lymph nodes (ICLN) in the neonatal phase. This study aimed to characterize age- and nutrition-related changes in the metabolically active bacterial and fungal composition of the ICLN in suckling and newly weaned piglets. Ten litters received only sow milk, while ten litters had access to creep feed from day of life (DoL) 10. Weaning occurred on DoL28. The ICLN were collected from 10 piglets/feeding group on each sampling day (DoL7, 14, 21, 28, 31 and 35) for RNA isolation, transcription into complementary DNA for 16 S rRNA and ITS2 amplicon sequencing. Age and weaning influenced the microbiome in the ICLN more than the nutrition during the suckling phase. Species richness and alpha-diversity of the bacterial but not fungal communities were increased on DoL7 and postweaning. Potential modes of action may have been linked to gut permeability at these ages and selective sampling by immune cells. Potential selective transfer of microbes may explain the dominance of Lactobacillus and Limosilactobacillus in the ICLN. Piglets that only drank sow milk comprised more Bacteroides in their ICLN on DoL35 compared to the creep fed piglets. Especially the role of fungi in the ICLN, including their mechanisms for translocation survival, needs further attention, as we detected metabolically active mold fungi and plant pathogens (e.g., Fusarium, Alternaria and Blumeria) in the ICLN.
Similar content being viewed by others


Introduction
The porcine gastrointestinal tract is quickly colonized after birth by a large variety of microbes originating from the vagina, nipple surface, and milk1,2. Concurrently, the gut barrier and immune tolerance develop, whereby the microbial colonization is a major driver for these maturational processes3,4. Commensal microbes translocate from the gut and accumulate in mesenteric lymphatic tissues, such as the ileocecal lymph nodes (ICLN), where they challenge and train the developing immune system5. The ICLN act as the border between the innate and adaptive branches of the local immune system, preventing microbes to reaching the systemic circulation6,7,8. However, both branches of the immune system are immature in newborn piglets9. Evidence of gut bacterial translocation into mesenteric lymph nodes exists for growing and finishing pigs10,11, whereas data for neonatal piglets and the presence of other microbial groups (e.g., fungi) in porcine ICLN are missing. Previously, we could show that Lactobacillus accumulated, probably by the action of immune cells, in the ICLN of 11-week-old pigs11. In this line of reasoning, dendritic cells and/or microfold (M) cells pass specific luminal microbes to the underlying lymphoid follicles to initiate an immune response12. It is not known whether a similar selective uptake of microbes takes place in the neonatal phase in piglets. While developmental processes continue during the suckling phase, weaning often interrupts this development, and is associated with dysbiosis, gut inflammation and an impaired gut barrier2,13. Consequently, more microbes may translocate into the ICLN in the days following weaning.
To mitigate weaning-associated gut disorders, creep feeding is routinely used in pig production systems as a nutritional strategy to prepare the digestive and secretory functions as well as the gut microbiome for the consumption of plant-based feed from the early suckling phase4. Theoretically, this dietary intervention should moderate the microbial translocation into the ICLN by supporting the barrier and immune functions in the first days after weaning, but scientific evidence is rare.
The objective of the present study was to characterize age-and nutrition-related changes in the bacterial and fungal communities present in the ICLN of suckling and newly weaned piglets. To study the effect of creep feeding, litters were grouped into litters that only drank sow milk and litters that received additional creep feed during the suckling period. Moreover, viable microbes may have a greater impact on the immune cells14; therefore, we isolated RNA from the ICLN to determine metabolically active bacterial and fungal taxa. Our study was based on the hypothesis that the bacterial and fungal numbers and diversity in the ICLN would be higher in the early suckling period and postweaning due to lower barrier functions at these ages. We also hypothesized that creep feeding would lead to lower abundances and diversity of microbes found in the ICLN postweaning.
Results
Data on growth performance and development of the microbiome in gastric and cecal digesta and feces and host physiology can be found in Lerch et al.4 and Metzler-Zebeli et al.15. The estimated creep feed intake of the piglets in the suckling phase can be found in Table S1.
Quantification of metabolically active bacteria and fungi by quantitative PCR
Across DoLs, the average log10 gene copies per gram lymph node tissue were 7.7 and 4.2 for bacteria (Fig. 1A) and fungi (Fig. 1B), respectively. The ANOVA indicated a trend (p = 0.090) for an age effect for bacterial 16 S rRNA gene copies and an age effect (p = 0.003) for the fungal 26 S rRNA gene copies. Accordingly, the total abundance of bacteria increased from DoL7 to DoL14 by 0.4 log units, whereas that of the fungi increased by 0.5 log units increased from DoL7 to DoL28 (p < 0.05). Creep feeding did not affect the total bacterial and fungal abundances.

Bacterial 16 S rRNA and fungal 26 S rRNA gene copies in ileo-cecal lymph nodes. (A) total bacteria, and (B) total fungi. One piglet group [sow milk group; n = 10 per day of life (DoL)] drank only sow milk during the suckling phase, the other piglet group [creep feed group; n = 10 per DoL] received additional creep feed from DoL10 until weaning which took place on DoL28. No creep feed effects and interactive effects of DoL and creep feed on total microbial abundances (p > 0.1).
Changes in bacterial and fungal diversity
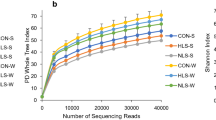
For the bacterial community, ICLN samples comprised on average 28,205 reads per sample. Bacterial species richness (Observed amplicon sequence variants (ASV)) and alpha-diversity (Shannon index) decreased from DoL7 to DoL14 (p < 0.05; Fig. 2). Postweaning, the bacterial species richness increased compared to DoL28 (p < 0.05; Fig. 2A), whereas both indices for the alpha-diversity, Shannon and Simpson, were only higher on DoL31 (p < 0.05) but not on DoL35 compared to DoL28 (Fig. 2B and C). For the fungal community, one sample contained on average 8,230 sequences. Age and nutrition did not influence the species richness and diversity of the fungal community throughout the experimental period. The permutational analysis of variance (PERMANOVA) based on Bray-Curtis dissimilarities supported an age effect (p < 0.01; Table S2) but no creep feed effect on the beta-diversity of both the bacterial and fungal communities. The age-related development of the beta-diversity for the bacterial and fungal communities was visualized in nonmetric multidimensional scaling (NMDS) ordination plots (Fig. 3A and B).

Species richness and alpha-diversity of bacterial (blue) and fungal (orange) communities in ileo-cecal lymph nodes. Age-related differences for (A) Observed amplicon sequence variant (ASV) index, (B) Shannon index, and (C) Simpson index in suckling and newly weaned piglets. One piglet group [sow milk group; n = 10 per day of life (DoL)] drank only sow milk during the suckling phase, the other piglet group [creep feed group; n = 10 per DoL] received additional creep feed from DoL10 until weaning which took place on DoL28. No creep feed effects and interactive effects of DoL and creep feed on diversity indices (p > 0.1).

Non-metric multidimensional scaling (NMDS) plot of pairwise Bray-Curtis dissimilarities. (A) Bacteria (stress level = 0.0802), and (B) fungi (stress level = 0.0910) in ileo-cecal lymph nodes of suckling and newly weaned piglets. Ellipses represent the standard deviation. Days of life are indicated by numbers (7, 14, 21, 28, 31 and 35) in the center of the ellipses.
Changes in bacterial taxonomy
The genus Lactobacillus dominated the bacteriome of the ICLN during the suckling and early postweaning phase with the highest relative abundance on DoL14 with 64.2% and lowest on DoL31 with 19.3% (adjusted p < 0.05; Fig. 4A; Table S3). The next most abundant taxa were Limosilactobacillus, Clostridium sensu stricto, Lactobacillaceae genus HT002 and Prevotella. The abundance of Limosilactobacillus decreased from DoL7 to DoL21, whereas it increased again on DoL28 before dropping a second time after weaning (adjusted p < 0.05). Clostridium sensu stricto and the Lactobacillaceae genus HT002 tended to increase from DoL7 to 28 (adjusted p = 0.066). Campylobacter, Ligilactobacillus, Blautia and the Oscillospiraceae genus UCG-005 were only detected in the ICLNs from DoL21 to DoL35. Further bacterial taxa that were among the 20 most abundant genera but at a relative abundance of < 2% included Bacteroides and Ruminococcus, as well as Escherichia-Shigella. During the suckling period, the 20 most abundant taxa covered about 84.9% of all reads. After weaning, this proportion dropped to 55.2% coverage of all reads, showing a diversification of the bacterial community. The most severe drop in abundances occurred for Lactobacillus, Limosilactobacillus from DoL28 to DoL31 and Lactobacillaceae HT002 from DoL28 to DoL35, which decreased by 60.6, 74.8 and 49.7%, respectively (adjusted p ≤ 0.05). Taxa among the 20 most abundant genera that dramatically increased in their relative abundances from DoL28 to DoL35 were Prevotella 9, Terrisporobacter, Romboutsia, Conexibacter, Colidextribacter (adjusted p < 0.05) and Streptococcus (adjusted p = 0.084), and from DoL28 to DoL31 Campylobacter, Blautia, Oscillobacteriaceae UCG-005 (adjusted p < 0.05). Alterations in the bacterial composition were also visible from DoL31 to DoL35 (Table S3). For instance, the relative abundance of Conexibacter and Colidextribacter increased (adjusted p < 0.05) and those of Ruminococcus and Lachnoclostridium tended (adjusted p < 0.10) to decrease from DoL31 to DoL35. Differential analysis showed that creep feeding had only minor effects on the bacteriome composition in the ICLN (Fig. 5A and B; Table S3). Bacteroides and the low abundant Prevotellaceae NK3B31 group were higher in piglets that drank only sow milk during the suckling phase compared to creep-fed animals but only on DoL35 (adjusted p < 0.05). Results on positive and negative correlations between the 20 most abundant bacterial genera and the abundance of the respective genera in cecal digesta on the various DoL can be found in Fig S1.

Relative abundances of the 20 most abundant bacterial and fungal genera in ileo-cecal lymph nodes. (A) Bacterial genera, and (B) fungal genera based on 16 S rRNA and ITS2 gene sequencing, respectively, in suckling and newly weaned piglets. One piglet group [sow milk group; n = 10 per day of life (DoL)] drank only sow milk during the suckling phase, the other piglet group [creep feed group; n = 10 per DoL] received additional creep feed from DoL10 until weaning which took place on DoL28. Effects (adjusted p < 0.05) and trends (adjusted p < 0.10) of DoL is indicated by ‘*’ and ‘#’, respectively.

Age- and creep feed-related effects on genera. Relative abundance of (A) Bacteroides, (B) Prevotellaceae NK3B31 group, (C) Alternaria, and (D) Trichoderma in the ileo-cecal lymph nodes of suckling and newly weaned piglets. One piglet group [sow milk group; n = 10 per day of life (DoL)] drank only sow milk during the suckling phase, the other piglet group (creep feed group; n = 10 per DoL) received additional creep feed from DoL10 until weaning which took place on DoL28. A-B represent bacterial genera, whereas C-D represent fungal genera.
Changes in fungal taxonomy
The 20 most abundant fungal genera covered 75.8% of all reads during the suckling phase and 82.8% on DoL31 and DoL35 (Fig. 4B; Table S4). Overall, there were large animal-to-animal variations for the various fungal genera that were present at an abundance > 0.05% of all reads. Except the trend (adjusted p = 0.070) for an unknown genus within Malasseziaceae which was more abundant on DoL31 compared to DoL28, the DoL did not affect the abundances of the 20 most abundant fungal genera. An unknown fungal genus (Incertae sedis), Fusarium and an unknown Chytridiomycota genus dominated the fungal community with relative abundances of 29.6, 11.0 and 10.2%, respectively, within the suckling period. The next most abundant genera comprised Hyphopichia, Starmerella, Aspergillus, Malassezia, Talaromyces and Saccharomyces with average abundances between 0.7 and 3.6% of all reads in the suckling period. Similar to the suckling period, the same fungal taxa continued to dominate the mycobiome postweaning on DoL31 and DoL35. Like for the bacterial genera, creep feeding had only a small effect on the fungal abundances (Fig. 5C-D; Table S4). The two genera that tended (fixed effect DoL, p < 0.10) to be different in creep fed piglets compared to piglets that only received sow milk on some of the sampling days were Alternaria and Trichoderma. Accordingly, differential analysis showed that Alternaria was generally less present in creep fed piglets compared to piglets that only drank sow milk (adjusted p < 0.094). Trichoderma, in turn, tended (adjusted p < 0.093) to be more abundant in creep fed piglets compared to piglets that only drank sow milk in the suckling and postweaning periods. The positive and negative correlations between the 20 most abundant fungal genera and the abundance of the respective genera in cecal digesta on the various DoL are presented in Fig S2.
Discussion
This study is one of the first surveys on the diversity and abundance of viable bacteria and fungi in the ICLN of suckling and newly weaned piglets. Our results demonstrate that age and weaning modified the translocation of microbial populations into the ICLN of neonatal piglets more than creep feeding. More specifically, age and weaning altered the metabolically active bacterial composition in the ICLN but less the metabolically active fungi. In this respect, our data show that the metabolically active community was richer in bacteria on DoL7 and postweaning, indicating an increased translocation of different taxa into the ICLN than on the other sampling days. The fact that the bacterial and fungal load (gene copies) did only increase by about 0.5 log units from DoL7 onwards may indicate that immunological control mechanisms were in place to control the transfer of microbes into the ICLN. Based on these findings, we can also speculate that the translocated metabolically active bacterial and fungal taxa played a vital role in the training of the immune system to become unresponsive towards the commensal gut microbiota and/or specific taxa. To support the latter assumption, the ICLN of the piglets were enriched with Lactobacillus and Limosilactobacillus, both taxa with beneficial capabilities for the host16. By contrast, in the case of pathobionts, the translocated microbes may trigger the development of chronic infections5, which needs more research. Some of the bacterial genera that include pathobionts (e.g., Clostridium perfringens within Clostridium sensu stricto) belong to the first colonizers of the porcine gut2, which probably explains their presence in the ICLN. Lastly, our data showed that creep feeding did not greatly change the translocation of microbes into the lymph nodes of the piglets neither during the suckling or postweaning period. Creep feeding only started on DoL10 and the amounts that the piglets consumed until DoL26 were small. This probably explains the small effect that creep feeding had on the microbial communities in the ICLN, corresponding to the observations made for the microbial communities in gastric and cecal digesta4.
The first collection of the ICLN took place on DoL7 in the present study, which should have been a few days after the gut closure occurring after birth2. In this respect, Prims et al.17 measured the volume densities of M cells in the ileal Peyer’s patches of neonatal piglets by which they could show that antigen sampling is possible from birth onwards. The authors related this ability to maternal lymphatic cells that originated from colostrum or transient milk and were found in neonatal lymphatic tissue17. Accordingly, maternal immune cells might have played a role for the microbial composition found in the ICLN of piglets on DoL7 in the present study. In this way, the maternal cells may have compensated the low functioning of the neonatal antigen presenting cells at this age18,19. Species richness and diversity in the intestinal lumen (ileum and cecum) show an opposite development to our findings for the ICLN, by being low at first and increasing until after weaning3,13. The question arises whether the greater species richness and diversity on DoL7 was intended for the priming of the immune system or still a sign for the permeability after birth for the uptake of maternal antigens. The same question can be asked for the sampling days after weaning when the increased species richness and diversity could have been a sign of a ‘leaky gut’ due to weaning or partly intended to train the immune system onto the novel bacterial composition in the gut. Three observations would support the latter assumption. First, the total bacterial load remained similar from pre- (DoL28) to postweaning phase. Second, the fungal composition and total numbers did not change much. Third, we observed a highly inflamed gut from the esophagus to the anus in the present piglets on DoL31, whereas on DoL35 the gut returned to its normal color4. Consequently, total gene copy numbers may suggest that the immune system ‘allowed’ or specifically subsampled a certain number of bacteria and fungi to be presented to the developing adaptive immune system in the ICLN, which needs further investigation. Previously, we studied the bacterial taxonomic composition in ICLN of young growing11 and finishing pigs5,10,20 but we missed to quantify their abundance to have data for comparison to support this assumption. Another possibility that may explain the higher species richness (DoL7 and postweaning) and diversity (postweaning) may be linked to the changes in taxa and hence to the motility (e.g., bacteria carrying flagella) and exopolymer structures (e.g. exopolysaccharides) on the cell surface, which may allow facilitated bacterial translocation through the gut epithelium21,22. From the relative abundances of the 20 most abundant bacterial genera which were more or less similar on DoL7 and DoL14, it can be deduced that the differences in species richness and diversity were mainly due to variation in low abundant taxa. Very little research has been performed in terms of the fungal colonization of piglet’s gut and immune organs; therefore, comparable data are missing. As mentioned before, the stability of the fungal composition suggest that a certain control may have been in place throughout the suckling phase but could have been also a reflection of gut mucosal fungal communities.
The bacterial communities in the ICLN of the suckling and early postweaning piglets in the present study differed from those that we found in older pigs10,11,20. The question that needs to be answered in future studies is whether this difference was mainly a sign for the maturational stage of the gut microbiome and immune system or whether environmental factors, such as diet and housing, and analytical factors related to the DNA extraction and 16S rRNA amplicon sequencing23 played a bigger role. Literature suggests that bacteria that reach the mesenteric lymph nodes in immune cells are kept alive for antigen presentation several days or weeks24,25. In order to do so, the microbes rely on nutrients provided by the immune cells or in the lymph, which needs to be shown for newborn piglets.
From the location of the ICLN in the plica ileocecalis, the detected bacterial and fungal taxa originated from the ileum, cecum and proximal colon. In accordance with that, the communities in the ICLN comprised bacterial and fungal genera that we also detected in cecal digesta4. Some taxa correlated in their abundances with their occurrence in cecal digesta on specific sampling days (e.g., Prevotella on DoL7). Other major genera in the ICLN, e.g., Lactobacillus, did not correlate to their cecal abundances, again demonstrating that factors, such as selective sampling by immune cells but also the different gut niches as sources of microbial origin and trans- and paracellular mucosal control of microbe transfer (e.g., via gap- and tight junctions and endocytosis)26, are involved in the microbial translocation. Regarding the latter, cell surface structures (e.g., glycocalyx) could have facilitated the translocation of Lactobacillaceae but this topic needs further research. Due to their proximity, it may be thinkable that the taxonomic composition in the ICLN of the neonatal piglets may represent the gut mucosal communities, which should be examined in a future study. Our previous research in young growing and finishing pigs has shown that this is not necessarily the case10,11. Similar to the ICLN in 11-week-old pigs11, the bacterial community in the ICLN of the neonatal piglets in the present study was enriched in Lactobacillaceae, mainly Lactobacillus and Limosilactobacillus. Likewise, an unclassified Lactobacillaceae genus (HT002) and Ligilactobacillus were among the 20 most abundant genera at all sampling time points. In the gut, Lactobacillaceae control the action and hinder their mucosal attachment of other bacteria including pathobionts via the production of antimicrobial peptides and organic acids27 and have immunomodulatory properties28. Due to their high dominance in the lymph nodes, it would be interesting to know whether Lactobacillus and related species exert similar effects in the ICLN and control the viability of other microbes, which could also explain their dominance. Moreover, sugar-fermenting Romboutsia29, as well as milk-glycan fermenting Bacteroides, Ruminococcus30 and Prevotella species (e.g., P. stercorea)31 were among the most abundant taxa, for which the cecum may have been the gut site of origin4. Likewise, first colonizers of piglet’s gut (e.g., Clostridium sensu stricto and Escherichia) were present at low to moderate abundance levels2,15,23. Similarly, Campylobacter is often found in the commensal gut microbiome of pigs15,32, which may explain their low abundance and their presentation to antigen producing cells in the ICLN.
The fungal community comprised several taxa that were not classified at genus level, which makes it difficult to make assumptions about their functional roles. Besides, the ICLN comprised taxa that are typically found on plants from the first sampling day (DoL7) onwards. Different origins of these fungi are probable including that the suckling piglets took them up from the environment or sow feces (e.g., Didymella and Blumeria)33,34. Other genera were skin- (e.g., Malasezziaceae) or gut-related fungi (e.g. Kazachstania), with the mother sow as source of origin33,34. The presence of several mold fungi (e.g., Fusarium, Penicillium, Alternaria and Aspergillus) among the 20 most abundant fungal taxa in the ICLN is concerning as they can cause chronic inflammation and may weaken the developing immune system35. Their origin was probably airborne from the barn environment, sow feed and/or sow feces33,34. However, it cannot be excluded that fungi were also taken up by the piglets via sow milk36 and then transferred to the ICLN, which should be investigated more closely in the future.
The composition of the bacterial and fungal taxa in creep fed piglets compared to sow milk only fed piglets was not as different as we would have expected, especially after weaning. Of special interest are the observations for certain bacterial (Bacteroides and Prevotellaceae NK3B31 genus) and fungal genera (Alternaria) among the most abundant taxa that showed diet-related differences after weaning but not in the suckling phase when the nutrition was different. This may point towards differences in the gut barrier and mucosal immune response towards the novel digesta composition (i.e., feed residuals and digestion products, microbes and microbial products), upregulating pro-inflammatory pathways and lowering mucosal barrier function13,34. The higher species richness and diversity would support this assumption for DoL31. However, Bacteroides, the unknown Prevotellaceae genus as well as Alternaria were enriched on DoL35 but not on DoL31. Also, we did not find corresponding differences in the jejunal and cecal expression of tight-junction protein genes4 to support lower mucosal barrier function. Therefore, it would be interesting to know whether the enrichment in these genera on DoL35 was related to gut mucosal abundances. The positive correlation between Bacteroides in the ICLN and cecal digesta postweaning may indicate that this may have been the case, whereas correlations between ICLN and cecal digesta for the Prevotellaceae genus, Saccharomyces and Alternaria with diet-related differences in the suckling or postweaning period were absent. Moreover, it is worth mentioning that the abundance of Bacteroides drastically dropped in gastric and cecal digesta on DoL31 due to the lack of milk glycans as substrate4, making it more difficult to explain the present finding. During the suckling phase Trichoderma that was enriched in the ICLN of creep fed piglets on DoL14 compared to sow milk only fed piglets may have translocated from the cecum as indicated by the present positive correlations.
In conclusion, the present results provide evidence for the transfer of viable bacteria and fungi into ICLN in neonatal piglets. Age and weaning had a stronger effect on the bacterial diversity than the nutrition, suggesting a role of the maturational stage of the gut barrier and specific immune cell activity. The role of fungi in the interaction with immune cells needs further investigation as well as the modes of action how microbes translocate and survive in ICLN of neonatal piglets, including the impact of maternal immune cells. In future studies, it is also worth to compare the microbial composition in the ICLN to the luminal and mucosal communities at all closely associated gut sites and niches to obtain a better understanding about the origin of the translocated microbes.
Materials and methods
Animals and housing
The study was conducted under practical production conditions at the pig facility of the University of Veterinary Medicine Vienna (VetFarm) and has been described in detail in Lerch et al.4 and Metzler-Zebeli et al.15. Only the parts of the animal management, handling and sampling are given here that are relevant for the present paper. Management and feeding of sows and piglets corresponded to the routine protocol at the pig facility. In two consecutive replicate batches, each lasting 35 days (from farrowing to one week post-weaning), the cross-bred progeny (Swiss Large White × Piétrain) of 20 Large White sows were used. Sows farrowed freely in individual BeFree pens (Schauer, Agrotonic, Prambachkirchen, Austria; 2.3 × 2.6 m) within 48 h. Piglets were weighed and identified with an ear tag after birth. The BeFree pens were equipped with a bowl drinker, feeder and hayrack for the sow and bowl drinkers and a nest with heated flooring for the piglets. Mainly small birth weight piglets were removed from the experiment on DoL1 to adjust litter size to a maximum of 13 piglets. The other piglets remained with their respective mother throughout lactation. Piglets were weaned on DoL28. After weaning, piglets were moved to the weaner pig room and kept in groups of a maximum of 20 animals (pen size 3.3 m × 4.6 m) by housing two to three litters of the same dietary group together. Pens were equipped with a piglet nest, nipple and bowl drinkers and one round feeder. Straw was provided as bedding material. Sows and piglets had always free access to water.
Feeding and dietary groups
Throughout lactation, sows were fed a commercial cereal-soybean meal-based lactation diet according to the regular feeding protocol at the pig facility (Table S4). The feed amount was gradually increased post-farrowing until an average amount of 8.7 kg feed per sow and day was reached. Litters were divided into two dietary groups which were balanced for the parity of the sow. Per replicate batch, piglets from five litters suckled only sow milk (sow milk group), whereas the piglets from the other five litters had additionally free access to creep feed from DoL10 (creep feed group). In total, there were 10 litters that only drank sow milk and 10 litters that had access to creep feed.
The creep feed was a commercial milk replacer (Table S4), which was freshly prepared from dry powder according to the manufacturer’s instructions at least twice daily. The dry powder of the milk replacer was mixed 1:5 (wt/vol; 200 g/L) with warm water (45 °C) to achieve a thin liquid. Then, the milk replacer cooled down and was fed at ambient temperature. Each litter received a minimum amount of 1000 mL per day (500 mL at 08:00 and 500 mL at 15:00 h) in special stainless steel feeders; more when piglets finished their portion. Sows had no access to the creep feed. Leftover creep feed and spills were collected and recorded, subsamples were taken for dry matter and nutrient analysis and the creep feed intake was estimated on litter basis daily4,15. On DoL24 and 25, the creep feed was gradually mixed with the prestarter diet (Supplementary Table S1) and fed as mash. From DoL26 until weaning, the piglets from the creep feed group were fed 100% of the prestarter diet in dry form. After weaning on DoL28, piglets had free access to the prestarter diet in dry form until the end of the experiment on DoL35. Litters in the sow milk group did not receive feed before weaning. They were offered the prestarter diet in dry form ad libitum from weaning (DoL28) to DoL35. All diets were commercial complete feeds and met the current recommendations for nutrient requirements37.
Collection of ileo-cecal lymph nodes
In each of the two replicate batches, we collected ICLN from 10 piglets (5 males and 5 females) each on DoL7, 14, 21, 28, 31 and 35. At each of the 6 sampling days, 10 piglets per dietary group were selected based on average body weight development across litters (n = 5/dietary group/sampling day/replicate batch). The selection of piglets on each sampling day and the consecutive sampling days was balanced for sex and litter.
The piglet was weighed and anaesthetized into the ear vein with azaperone (Stresnil 40 mg/mL, Elanco Tiergesundheit AG, Basel, Switzerland) and ketamine (Narketan 100 mg/mL, Vetoquinol Österreich GmbH, Vienna, Austria), and euthanized with intracardiac application of embutramide (T61, Intervet GesmbH, Vienna, Austria) after complete sedation was ensured. The piglet was bled, the abdomen opened and the whole gut removed aseptically. Individual gut segments were sorted, clamped and the ICLN were identified. The ICLNs were collected near the ileo-colic artery caudal to the cecum and immediately placed into ice-cold phosphate-buffered saline. ICLNs were disinfected by flaming before removing surrounding tissues (i.e. fat and connective tissues), cutting the lymph nodes into tiny pieces and snap-freezing them in liquid nitrogen.
RNA extraction and targeted quantitative PCR
Total RNA was extracted from 20 mg of ICLN tissue using the RNeasy Mini Qiacube kit (Qiagen, Hilden, Germany)11. The ICLN sample was combined with 350 μl lysis buffer (Qiagen) and 0.6 g ceramic beads (diameter [Ø], 1.4 mm). Samples were homogenized for 30 s (6.5 m/s) using the FastPrep-24 instrument (MP Biomedicals, Heidelberg, Germany). The remaining procedures were performed on the Qiacube robotic workstation (Qiagen, Hilden, Germany) according to the regular protocol of the kit. DNAase I (Invitrogen TURBO DNA-free Kit; Thermo Fisher Scientific Inc., Waltham, MA, USA) was used to remove genomic DNA. The Qubit 4 fluorometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) with the Qubit RNA assay kit were used to quantify the total RNA concentration. Afterwards, 2 μg complementary DNA (cDNA) were synthesized from the respective amount of RNA using a high-capacity reverse transcription kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). In addition, 0.5 μl RNase inhibitor (Biozym, Hessisch Oldendorf, Germany) was added to each reaction mixture.
First, total bacterial and fungal abundances were quantified using a qTower384 qPCR System (Analytik Jena, Jena, Germany), similar to the protocol described in Lerch et al.4. The utilized primer sets for total bacteria and fungi quantification were published before (Table S5). The pipetting of the reactions was conducted by means of a robot (epMOTION 5075 TMX, Eppendorf SE, Hamburg, Germany). Each standard and sample reaction (10 μl) comprised 7 μl master mix including innuMIX qPCR DS Green Standard (IST Innuscreen, Berlin, Germany), forward and reverse primers (concentration in reaction: 100 nM) and 25 ng cDNA template. After an initial denaturation step at 95 °C for 2 min, 40 cycles of 95 °C for 30 s followed with primer annealing and elongation at 60 °C for 30 s were completed. Fluorescence was measured in all cycles. To verify the specificity of the PCR product, melting curve analysis was performed. Negative template controls were run in triplicates on each plate, whereas samples and the serial dilutions of the standards were run in triplicates. Standards were 10-fold serial dilutions (107 to 103 molecules/μl) of the purified and quantified PCR products using pooled DNA from the ICLN samples from this study. The final copy numbers were calculated using the equation: (QM × C × DV)/ (S×V), where QM is the quantitative mean of the copy number, C is the RNA concentration of each sample, DV is the elution volume of isolated RNA, S is the cDNA amount (ng) and V is the weight of the sample (g) subjected to RNA isolation. Amplification efficiencies (E = 10(−1/slope)) and coefficient of determination (linearity) are provided in Table S5.
Sequencing and bioinformatics
To disentangle the age- and nutrition-related effects on the bacterial and fungal composition in the ICLN of the piglets, the V3-V4 hypervariable regions of the bacterial 16 S rRNA gene and the ITS2 region of fungi were amplified. The primer systems were 341 F-ill (5′-CCTACGGGNGGCWGCAG-3′) and 806R-ill (5′-GACTACHVGGGTATCTAATCC-3′) for the V3-V4 region of the 16 S rRNA gene and ITS3 (5′-GCATCGATGAAGAACGCAGC-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) for the ITS2 region. The same cDNA samples that were used for total microbial quantification were sent for library preparation (NEBNext Ultra II DNA Library Prep Kit, Illumina, San Diego, CA, USA) and Illumina sequencing at a commercial provider (Novogene). Equimolar pools of samples were sequenced to generate 250 bp paired-end raw reads in the Novaseq 6000 platform (Illumina). Demultiplexing and trimming of the raw sequences was performed by Novogene. Raw sequencing reads (Fastq files) of the 16 S rRNA and fungal ITS2 amplicons were independently processed, aligned and categorized using the Divisive Amplicon Denoising Algorithm 2 (DADA2; version 1.26.0)38 in R studio (version 1.4.1106). Sequences were pre-filtered and reads with ambiguous bases were removed using the ‘filterAndTrim’ function. For the bacterial 16 S rRNA amplicons, the first 10 nucleotides were trimmed and the total length of forward and reverse reads were truncated to 220 nucleotides to account for the decrease in quality score of the following nucleotides using the ‘filterAndTrim’ function. For the fungal ITS2 amplicons, the first 10 nucleotides were trimmed to account for the decrease in quality score of the following nucleotides. Additionally, a minimum length of 50 nucleotides was enforced to remove very low length sequences using the ‘filterAndTrim’ function. Reads exceeding the probabilistic estimated error of two nucleotides were removed in the same step (‘filterAndTrim’ function). After de-replication of the filtered data and estimation of error rates, ASV were inferred. Thereafter, the function ‘mergepairs’ was used to merge the denoised pairs of forward and reverse reads, rejecting any pairs which did not sufficiently overlap, or which contained too many mismatches in the overlap region. Chimera were removed using the ‘removeBimeraDenovo() function’. Taxonomy was assigned using the function ‘assignTaxonomy’, which implements the RDP Naive Bayesian Classifier algorithm and reference databases - SILVA 138.1 ribosomal RNA (rRNA) database for bacteria39 and UNITE ITS database (version 9.0; release date 2022-11-29) for fungi40.
Taxonomic tables, taxonomic assignment and corresponding metadata were combined to create phyloseq objects in the R phyloseq (version 1.42.0)41. The raw sequence counts from the taxa tables were collapsed and compositionally normalized such that each sample summed to 1. The relative abundances at the genus rank were statistically analyzed as described below. The alpha diversity metrics were calculated using the phyloseq “estimate_richness” function from the rarefied amplicon sequence variants tables. From the diversity metrics, only the Observed ASV index, the Simpson and the Shannon diversity measures were subjected to statistical analysis. For beta-diversity, NMDS ordination plots based on the Bray-Curtis dissimilarity matrix (‘metaMDS’ function; vegan R package (version 2.6.4) were created to examine microbiome clustering by age and nutrition during the suckling phase. In addition, PERMANOVA was performed on the Bray–Curtis dissimilarity matrix using the adonis2 function to evaluate associations between beta-diversity and days of life and nutrition, separately for bacteria and fungi. Statistical significance was calculated after 999 random permutations. At genus level, the raw read counts from the various taxa, separately for the bacterial and fungal reads, were collapsed and compositionally normalized such that each sample sums to 1.
Statistical and multivariate analyses
Bacterial (> 0.2% relative abundance of all reads) and fungal genera (> 0.05% relative abundance of all reads) were statistically analyzed. The ranked relative abundances were analyzed in SAS. Normal distribution of the residuals of the data for the total and relative bacterial and fungal abundances in the ICLN was evaluated using the Shapiro-Wilk Test and UNIVARIATE procedure in SAS (version 9.4; SAS Stat Inc., Cary, NC, USA). Thereafter, the total and compositional abundances of bacteria and fungi (dependent variable) and alpha-diversity indices were subjected to analysis of variance using the MIXED procedure in SAS and repeated measures to investigate effects with increasing age of the piglets. We performed an analysis of variance in order to be capable to consider important covariates when conducting an experiment with piglets from different litters and sexes over the course of two replicate batches. Fixed effects included sex, replicate batch, DoL, feeding group (sow milk only versus additional creep feed during the suckling phase), litter and the respective two- and three-way interactions. The piglet represented the experimental unit. The Kenward Roger method (dffm = kr) was used to approximate degrees of freedom. The probability of difference option in SAS was used to perform pairwise comparisons among least squares means. We applied the Bonferroni correction to adjust the raw P values for the relative abundances of bacterial and fungal genera42. Data were expressed as least squares means ± standard error of the mean and differences with p ≤ 0.05 and 0.05 < p ≤ 0.1 were considered as significant and trend, respectively. Differences between sexes were negligible. Pearson correlation analysis was performed for the 20 most abundant bacterial and fungal genera occurring in the ICLN with the abundance of the respective taxa in cecal digesta, if present, using PROC CORR in SAS. Correlations were calculated separately for the various DoLs. For data of the cecal bacterial community, all DoLs were available, whereas the fungal community in cecal digesta was only determined on DoL14, 28, 31 and 354. Descriptive statistics for the creep feed intake of piglets during the suckling period were performed using PROC MEANS in SAS4.
Data availability
The raw sequencing data employed in this article has been submitted to the NCBI’s sequence read archive (https://www.ncbi.nlm.nih.gov/sra); BioProject: PRJNA1050750 (bacteria) and PRJNA1051997 (fungi).
Abbreviations
- cDNA:
-
Complementary DNA
- DADA2:
-
Divisive amplicon denoising algorithm
- DoL:
-
Day of life
- ICLN:
-
Ileo-ceca lymph nodes
- ITS:
-
Internal transcribed spacer
- NMDS:
-
Non-metric multidimensional scaling
- PERMANOVA:
-
Permutational multivariate analysis of variance
- rRNA:
-
Ribosome-ribonucleic acid
References
Guevarra, R. B. et al. Piglet gut microbial shifts early in life: causes and effects. J. Anim. Sci. Biotechnol. 10, 1 (2019). https://doi.org/10.1186/s40104-018-0308-3
Metzler-Zebeli, B. U. Porcine gut microbiota and host interactions during the transition from the suckling to post-weaning phase. In: Gut Microbiota, Immunity, and Health in Production Animals, (eds Kogut, M. H. & Zhang, G.) Springer International Publishing: Cham, Switzerland, https://doi.org/10.1007/978-3-030-90303-9. (2022).
Arnaud, A. P. et al. Post-natal co-development of the microbiota and gut barrier function follows different paths in the small and large intestine in piglets. FASEB J. 34, 1430–1446 (2020). https://doi.org/10.1096/fj.201902514R
Lerch, F. et al. An insight into the temporal dynamics in the gut microbiome, metabolite signaling, immune response, and barrier function in suckling and weaned piglets under production conditions. Front. Vet. Sci. 10, 1184277 (2023). https://doi.org/10.3389/fvets.2023.1184277
Mann, E., Dzieciol, M., Metzler-Zebeli, B. U., Wagner, M. & Schmitz-Esser, S. Microbiomes of unreactive and pathologically altered ileocecal lymph nodes of slaughter pigs. Appl. Environ. Microbiol. 80 (1), 193–203 (2014). https://doi.org/10.1128/AEM.03089-13
Davies, P. R. Intensive swine production and pork safety. Foodborne Pathog Dis. 8, 189–201 (2011). https://doi.org/10.1089/fpd.2010.0717
Macpherson, A. J. & Smith, K. Mesenteric lymph nodes at the center of immune anatomy. J. Exp. Med. 203, 497–500 (2006). https://doi.org/10.1084/jem.20060227
von Andrian, U. H. & Mempel, T. R. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 3, 867–878 (2003). https://doi.org/10.1038/nri1222
Stokes, C. R. The development and role of microbial-host interactions in gut mucosal immune development. J. Anim. Sci. Biotechnol. 8, 12 (2017). https://doi.org/10.1186/s40104-016-0138-0
Zwirzitz, B. et al. Microbiota of the gut-lymph Node Axis: depletion of Mucosa-Associated Segmented filamentous Bacteria and Enrichment of Methanobrevibacter by Colistin Sulfate and Linco-Spectin in pigs. Front. Microbiol. 10, 599. https://doi.org/10.3389/fmicb.2019.00599 (2019). Erratum in: Front Microbiol. 2020;11:1051.
Klinsoda, J., Vötterl, J., Zebeli, Q. & Metzler-Zebeli, B. U. Alterations of the viable Ileal Microbiota of the Gut Mucosa-Lymph Node Axis in Pigs Fed phytase and lactic acid-treated cereals. Appl. Environ. Microbiol. 86 (4), e02128–e02119 (2020). https://doi.org/10.1128/AEM.02128-19
Kobayashi, N., Takahashi, D., Takano, S., Kimura, S. & Hase, K. The roles of Peyer’s patches and Microfold cells in the Gut Immune System: relevance to Autoimmune diseases. Front. Immunol. 10, 2345 (2019). https://doi.org/10.3389/fimmu.2019.02345
Lerch, F. et al. Exposure to plant-oriented microbiome altered jejunal and colonic innate immune response and barrier function more strongly in suckling than in weaned piglets. J. Anim. Sci. 100 (11), skac310 (2022). https://doi.org/10.1093/jas/skac310
Mann, E. et al. Psychrophile spoilers dominate the bacterial microbiome in musculature samples of slaughter pigs. Meat Sci. 117, 36–40 (2016). https://doi.org/10.1016/j.meatsci.2016.02.034
Metzler-Zebeli, B. U. et al. Temporal Microbial dynamics in feces discriminate by Nutrition, Fecal Color, consistency and sample type in Suckling and newly weaned piglets. Anim. (Basel). 13 (14), 2251 (2023). https://doi.org/10.3390/ani13142251
Ali, M. S. et al. Probiotics and Postbiotics as an alternative to antibiotics: an emphasis on pigs. Pathogens 12 (7), 874 (2023). https://doi.org/10.3390/pathogens12070874
Prims, S. et al. Effect of artificial rearing of piglets on the volume densities of M cells in the tonsils of the soft palate and Ileal Peyer’s patches. Vet. Immunol. Immunopathol. 184, 1–7 (2017). https://doi.org/10.1016/j.vetimm.2016.12.009
Williams, P. P. Immunomodulating effects of intestinal maternal colostral leukocytes by neonatal pigs. Can. J. Vet. Res. 57, 1–8 (1993).
Bandrick, M., Ariza-Nieto, C., Baidoo, S. K. & Molitor, T. W. Colostral antibody-mediated and cell-mediated immunity contributes to innate and antigen-specific immunity in piglets. Dev. Comp. Immunol. 43 (1), 114–120 (2014). https://doi.org/10.1016/j.dci.2013.11.005
Mann, E. et al. The metabolically active bacterial microbiome of Tonsils and Mandibular Lymph nodes of Slaughter pigs. Front. Microbiol. 6, 1362 (2015). https://doi.org/10.3389/fmicb.2015.01362
Ramos, Y. et al. Remodeling of the Enterococcal Cell Envelope during Surface Penetration promotes intrinsic resistance to stress. mBio 13 (6), e0229422 (2022). https://doi.org/10.1128/mbio.02294-22
Ramos, Y. et al. PolyGlcNAc-containing exopolymers enable surface penetration by non-motile Enterococcus faecalis. PLoS Pathog. 15 (2), e1007571 (2019). https://doi.org/10.1371/journal.ppat.1007571
Dong, W., Ricker, N., Holman, D. B. & Johnson, T. A. Meta-analysis reveals the predictable dynamic development of the gut microbiota in commercial pigs. Microbiol. Spectr. e0172223 (2023). https://doi.org/10.1128/spectrum.01722-23
Nagl, M. et al. Phagocytosis and killing of bacteria by professional phagocytes and dendritic cells. Clin. Diagn. Lab. Immunol. 9 (6), 1165–1168 (2002). https://doi.org/10.1128/cdli.9.6.1165-1168.2002
Hooper, L. V., Littman, D. R. & Macpherson, A. J. Interactions between the microbiota and the immune system. Science. 336 (6086), 1268–1273 (2012). https://doi.org/10.1126/science.1223490
Morikawa, M. et al. Microbiota of the small intestine is selectively engulfed by phagocytes of the Lamina Propria and Peyer’s patches. PLoS One. 11 (10), e0163607 (2016). https://doi.org/10.1371/journal.pone.0163607
Walter, J. Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl. Environ. Microbiol. 74 (16), 4985–4996 (2008). https://doi.org/10.1128/AEM.00753-08
Li, B., Pan, L. L. & Sun, J. Novel probiotic lactic acid Bacteria were identified from healthy infant feces and exhibited anti-inflammatory capacities. Antioxid. (Basel). 11 (7), 1246 (2022). https://doi.org/10.3390/antiox11071246
Gerritsen, J. et al. Romboutsia hominis sp. nov., the first human gut-derived representative of the genus Romboutsia, isolated from ileostoma effluent. Int. J. Syst. Evol. Microbiol. 68 (11), 3479–3486 (2018). https://doi.org/10.1099/ijsem.0.003012
Salcedo, J., Frese, S. A., Mills, D. A. & Barile, D. Characterization of porcine milk oligosaccharides during early lactation and their relation to the fecal microbiome. J. Dairy. Sci. 99 (10), 7733–7743 (2016). https://doi.org/10.3168/jds.2016-10966
Hayashi, H., Shibata, K., Sakamoto, M., Tomita, S. & Benno, Y. Prevotella copri sp. nov. and Prevotella stercorea sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 57 (Pt 5), 941–946 (2007). https://doi.org/10.1099/ijs.0.64778-0
Holman, D. B., Brunelle, B. W., Trachsel, J. & Allen, H. K. Meta-analysis to define a core microbiota in the Swine Gut. mSystems. 2 (3), e00004–17 (2017). https://doi.org/10.1128/mSystems.00004-17
Summers, K. L., Frey, J. F., Ramsay, T. G. & Arfken, A. M. The piglet mycobiome during the weaning transition: a pilot study1. J. Anim. Sci. 97 (7), 2889–2900 (2019). https://doi.org/10.1093/jas/skz182
Arfken, A. M., Frey, J. F. & Summers, K. L. Temporal dynamics of the gut bacteriome and mycobiome in the weanling pig. Microorganisms. 8, 868 (2020). https://doi.org/10.3390/microorganisms8060868
Wang, L. et al. Gut mycobiome and metabolic diseases: the known, the unknown, and the future. Pharmacol. Res. 193, 106807 (2023). https://doi.org/10.1016/j.phrs.2023.106807
Alonso, V. A., Velasco Manini, M. A., Pena, G. A. & Cavaglieri, L. R. Fist report on fumagillin production by aspergillus fumigatus sensu stricto gliotoxigenic strains recovered from raw cow milk and clinical samples in Argentina. Rev. Argent. Microbiol. 54 (3), 243–246 (2022). https://doi.org/10.1016/j.ram.2022.03.003
National Research Council (NRC). Nutrient Requirements of Swine 10th edn (National Academy, 2012).
Callahan, B. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Meth. 13, 581–583 (2016). https://doi.org/10.1038/nmeth.3869
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013). https://doi.org/10.1093/nar/gks1219
Nilsson, R. H. et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47 (D1), D259–D264 (2019). https://doi.org/10.1093/nar/gky1022
McMurdie, P. J. & Holmes, S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 8, e61217 (2013). https://doi.org/10.1371/journal.pone.0061217
Lin, H. & Peddada, S. D. Analysis of microbial compositions: a review of normalization and differential abundance analysis. NPJ Biofilms Microbiomes. 6, 60 (2020). https://doi.org/10.1038/s41522-020-00160-w
Acknowledgements
The authors thank D. Verhovsek, S. Posseth, T. Strini (Vetfarm), and T. Enzinger (Centre for Animal Nutrition and Welfare) for their excellent help with animal caretaking, assistance during sampling.
Funding
This research was funded by the Austrian Federal Ministry for Digital and Economic Affairs and the National Foundation for Research, Technology and Development. The Christian Doppler Laboratory for Innovative Gut Health Concepts of Livestock is financially supported by BIOMIN Holding GmbH, which is part of dsm-firmenich.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiment: BMZ. Animal experiment: FL, FY, JV, DV. Sample collection: JK, FL, JV, SK, BMZ. Data analysis: SK, BMZ. Writing-Original Draft Preparation: BMZ. Writing-Review & Editing: JK, FL, FY, and JV. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
All procedures involving animal handling and treatment were approved by the institutional ethics committee of the University of Veterinary Medicine Vienna and the National authority according to the Law for Animal Experiments, Tierversuchsgesetz (BMWFW- 68.205/00936- V3b/2019). The pig experiment was performed in accordance with the university and national regulations and complied to the ARRIVE guidelines.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Metzler-Zebeli, B.U., Lerch, F., Yosi, F. et al. Temporal dynamics in the composition of metabolically active bacteria and fungi in the ileo-cecal lymph nodes of suckling and newly weaned piglets. Sci Rep 14, 30902 (2024). https://doi.org/10.1038/s41598-024-81227-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-81227-y
