
Abstract
Filamentous Fungal Human Pathogens (FFHPs) such as Aspergillus fumigatus, are growing resistant to currently available antifungal drugs. One possible target, the Nucleoside diphosphate kinase (Ndk) is significant for nucleotide biosynthesis and crucial for fungal metabolism. Violacein, a natural food colorant, was examined for its antifungal effects against Aspergillus fumigatus via computational approach against the Ndk protein. Known and predicted interactions of Ndk with proteins was performed using the STRING application. Molecular docking was performed using Schrodinger Maestro software (V.14.1) under enhanced precision docking, with OPLS4 forcefield. MDS was performed for 500ns under OPLS4 forcefield and the TIP3P solvent system. The geometry optimization for DFT was performed using the Becke 3-parameter exchange functional (B3LYP) method. The Molecular Docking Studies revealed significant interactions with good binding energy between Violacein and Ndk. Subsequent MD Simulations confirmed the stability of Violacein-Ndk complex, compared to the reference ligand-complex, indicating a stable interaction between the protein and violacein. The energy band gap of violacein was found to be 0.072567 eV suggesting its softness with lower kinetic stability and higher chemical reactivity. The results suggest Violacein could potentially disrupt nucleotide metabolism by targeting Ndk, thus demonstrating antifungal activity. However, further experimental validation is required to confirm these computational findings and explore the practical use of Violacein in antifungal treatments.
Similar content being viewed by others
Introduction
Drug-resistant fungal infections are becoming increasingly prevalent, which exacerbates patient conditions while rendering treatment more challenging1. Since invasive fungal infections frequently have 50% or greater mortality rates, the presence of antifungal resistance, if any, increases the likelihood of worsening the medical condition because of the challenges in understanding the resistance mechanism and the availability of limited therapeutic options2,3. Antifungal resistance, which includes both novel resistant versions of previously vulnerable disease-causing fungi such as the Aspergillus fumigatus (A. fumigatus) and a whole new emerging species, the yeast Candida auris that are resistant to various antifungals, is a growing concern universally3,4,5. Accessibility to susceptibility testing and increased usage of antifungal drugs has led to increased knowledge and reports of resistance in Aspergillus infections6. A. fumigatus is a widespread saprophytic and thermophilic ascomycetes soil-borne fungus found often growing on food and decaying organic matter7. The fungus is thermo-tolerant, exhibiting growth from 12 to 65 °C, optimal at 35 °C. The surface of the fungus is covered with millions of small grey-green conidia (2–3 μm in diameter), produced in chains on separate phialides emerging from conidiophores borne on characteristic conidial heads. The conidia can rapidly get airborne, aiding in dispersal8,9, making them one among the commonly encountered air spora in indoor and outdoor environments, including agricultural environments or homes. The fungus can cause numerous grave ailments in both immunocompetent and immunocompromised patients10. Reportedly, it has been identified to cause the most severe diseases compared to other Filamentous Fungal Human Pathogens (FFHPs) from food11,12. Patients are at high risk of Aspergillus infections if consumed with foods contaminated by the fungi13,14. A. fumigatus is also one of the primary causes of the development of invasive aspergillosis, leading to fatalities in patients with hematological malignancies, those undergoing chemotherapy, and recipients of hematopoietic cell transplants15. The most common site of hematogenic spread is the central nervous system, leading to brain abscess, stroke, or, less frequently, meningitis, and associated with devastating mortality rates16. Approximately 85–90% of cases of invasive aspergillosis are reportedly caused by A. fumigatus, making it the cause of the most dangerous infections compared to others17. Including A. fumigatus infections in the crucial antimicrobial resistance danger list released by the US CDC in 2019 has formally acknowledged the growing public health burden18. Synthetic drugs termed azoles have been used extensively in therapy. Despite their effectiveness, there have been reports of azole-resistant pathogens owing to their augmented use. The most comprehensive study on A. fumigatus antifungal susceptibility testing discovered its resistance towards fluconazole and ketoconazole indicating the resistance pattern towards azole group of compounds. Hence, the quest for novel and safer alternative medicines from other natural resources (plants/microbes) as drug leads is being revitalized particularly in tackling anti-microbial resistance.
With a second-highest prevalence only to A. flavus, this deadly human pathogen can be found in baked goods, beans, beverages, and chocolates; staple cereals like maize, rice, barley, and wheat; dairy; fruits; herbs and spices; meat, fish, and eggs; nuts, seeds, and vegetables. A. fumigatus has also been identified as a cause of postharvest disease and losses in a variety of food items including, cabbage, bean, onion, garlic, potato and guava in various marketplaces throughout the world19,20. The mechanism of pathogenicity is attributed to the production of virulence factor, Gliotoxin leading to immune suppression, necrosis, structural alterations in tight junctions, and neuronal damage by impairing the human blood-brain barrier via cytotoxicity towards the astrocytes. Additionally, the fungus elaborates Fumagillin, a mycotoxin that inhibits neutrophil functions, causes eryptosis, and damages lung epithelia. Given the potential hazard of A. fumigatus contamination in food, there is a mounting necessity for food-grade antifungal drugs to address both public health and food security concerns.
Bacterial secondary metabolites have gained attention as natural therapeutic agents in treating various ailments, including allergies, autoimmune disorders, microbial infections, cancers, cardiac and neurological disorders21. Multiple instances have demonstrated the effectiveness of secondary metabolites originating from microorganisms as antifungal agents. Pyrrolnitrin from Pseudomonas, Serratia, and Burkholderia, utilized as an antifungal agent in the 1960s, displayed strong inhibition of the respiratory electron transport system in Saccharomyces cerevisiae, Penicillium atrovenetum, and P. oxalicum22. Similarly, 4-methylmellein, 4-hydroxymellein, 6-hydroxymellein, and tyrosol from Penicillium sp. were demonstrated to inhibit Fusarium oxysporum (F. oxysporum), A. flavus, and the yeast C. albicans23. Bacilysocin produced by Bacillus subtilis 168 has been shown to act against C. albicans24. Bacterial pigments are yet another class of secondary metabolites gaining popularity as natural therapeutics with their demonstrable biomedical efficacies on various human disorders including cancer, inflammation, immune responses, and anti-microbial effects. For instance, prodigiosin from Serratia marcescens inhibited several plant pathogenic fungi including Pythium myriotylum, Rhizoctonia solani, Phytophthora infestans, Sclerotium rolfsii, and F. oxysporum25. Pyocyanin isolated from Pseudomonas aeruginosa was reported to inhibit the growth and spore formation in F. oxysporum26. A yellow pigment, produced by Bacillus sp was found to inhibit the growth of R. solani, F. oxysporum, and S. rolfsii27.
Correspondingly, violacein, a bisindole purple-colored bacterial pigment has attracted research attention for its diverse biological functions including anti-bacterial, viricidal, antitumoral, anti-inflammatory, antifungal, antiprotozoan, antioxidant, immune-stimulatory, and ulcer protective potentials28,29. The pigment being less toxic, is effective against human and phytopathogenic fungi, and has applications in cosmetic and textile industries and dying medicinal fabrics. Additionally, violacein and deoxyviolacein have successfully demonstrated to be effective antiviral agents against the reverse transcriptase of human immunodeficiency virus 1 and the spike protein of SARS-CoV‑2 via computational and in vitro approaches30. Numerous plant and human pathogenic fungi, including Cryptococcus gastricus, R. solani, A. flavus, F. oxysporum, P. expansum, Trichophyton rubrum and C. albicans, were found susceptible to violacein31. Furthermore, the fungus Batrachochytrium dendrobatidis, the leading cause of the global reduction in frog populations, also showed susceptibility to violacein’s action32. Violacein has also been implicated in treating systemic infections caused by C. albicans and A. niger, limiting fungal growth, preventing the development of infectious fungemia, and lowering death rates28. These results highlight the beneficial properties of violacein as an antimicrobial agent, enabling further investigations into its antimicrobial potentials. Besides, violacein is also a desirable compound finding potential applications in the food industry as natural colourant33. The spray-dried powder of violacein was efficiently utilized in coloring food products like yogurt and jelly34. The concept of employing violacein, a food colorant, to prevent the food-borne A. fumigatus stems from applying the pigment in food for its proven anti-fungal properties. Through the bioinformatics approach that provides a platform for drug designing, much can be explored due to the precision and accuracy provided and guided by the knowledge of new chemical structures, functions, and targets. Since 1980, the concept of “rational use of pharmaceuticals” (also known as “structure-guided drug design”) has gained popularity. This involves the creation of target proteins and small molecules in silico as the basis for novel medication development35. Inverse docking, a novel technique for identifying therapeutic targets for medicines, enables the ligand to be assessed in silico for its binding to many possible targets in an electronic protein database. Thus, the bioinformatic approach can be utilized to determine the ligand-target interactions, thereby enabling insights into the mechanism of drug action. The present work is aimed at evaluating the efficacy of violacein as an antifungal agent, particularly against the Nucleoside Diphosphate Kinase (Ndk) protein of A. fumigatus via the in silico approach through molecular docking and molecular dynamic simulations.
Results
STRING analysis
The Nucleoside diphosphate kinase interaction network showed 11 nodes and 49 edges. The nodes indicated 11 proteins interacting with Ndk, while the 49 edges corresponded to the number of interactions or the predicted functional associations (Fig. 1).
Putative proteins interacting with Ndk were found to be:
-
Thymidylate kinase, known to catalyse the phosphorylation of thymidine 5′-monophosphate to thymidine 5′-diphosphate in the presence of ATP and magnesium, which is then converted to thymidine 5′-triphosphate by Ndk36.
-
Uridylate kinase, reported to catalyse the reversible transfer of the gamma-phosphoryl group from an ATP donor to Uridine monophosphate (UMP), yielding uridine diphosphate (UDP), which is the starting point for the synthesis of all other pyrimidine nucleotides36.
-
Ctp synthase (Cytidine triphosphate synthase), an enzyme involved in pyrimidine biosynthesis facilitating the conversion of uridine 5′-triphosphate (UTP) into cytidine 5′-triphosphate (CTP), the final step of cytidine nucleotides production37.
-
Deoxyuridine 5′-triphosphate pyrophosphatase, DUTPase (Dut), known to catalyse the hydrolysis of Deoxyuridine Triphosphate (dUTP) to form Deoxyuridine monophosphate, (dUMP) and pyrophosphate thereby eliminating dUTP and preventing its utilization by DNA polymerases during replication and repair38.
-
Guanylate kinase, catalyst of the ATP-dependent phosphorylation of GMP into GDP, and therefore synthesis of GTP39.
-
Ribonucleoside-diphosphate reductase subunits m1 and m2, a key enzyme in the synthesis of deoxyribonucleotides from ribonucleotides40.
-
adk1 and 2-involved in the metabolic monitoring of cellular adenine nucleotide homeostasis41.
-
Hemoglobin and proliferation-regulated protein, a nucleoside monophosphate kinase, catalysing the reversible phosphotransferase reaction between nucleoside triphosphates and monophosphates, i.e., monophosphates are converted to their corresponding diphosphate form42.

Interaction analysis of Ndk with other proteins via STRING.
Molecular docking
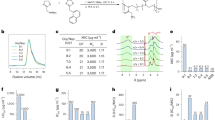
Interaction analysis of violacein with 6XPS revealed four hydrogen bonds with the positively charged amino acid residues ARG87, ARG104, and polar residues THR93, and ASN114, two salt bridges with the positively charged residuesLYS11 and ARG104, and two Pi-Pi Stalking interactions [with the hydrophobic amino acid residue PHE59] (Fig. 2). A glide g-score of − 7.211 kcal/mol was observed for the same interaction. The reference molecule (GDP) subjected to molecular docking with its parent protein generated a complex with glide g-score of − 7.010 Kcal/mol, and amino acid interactions including 4 hydrogen bonds (ASN114, THR93, HIE117, GLY118), and 2 Pi–Pi stalking interactions (PHE59) (Fig. 2). The reference molecule was superimposed upon itself before and after docking for docking protocol validation, and the RMSD was calculated. The superposition resulted in an RMSD of 1.54 Å. (Fig. 3).

2D interaction diagram of Violacein and reference ligand with Ndk protein.

Superposition of reference ligand (GDP) before and after molecular docking; The conformation of ligand before docking is coloured in blue and the conformation after docking is coloured in red.
Molecular dynamics simulation
To study the dynamics variation in the Cα backbone of the violacein-, reference molecule–protein complexes, and determine the stability of the selected protein, protein RMSD was calculated. Analysis of the simulation interaction diagram (Violacein-6XPS complex) revealed a fairly stable protein (Fig. 4a,c). In the first 200 ns, the protein RMSD was seen to be stabilized between 2 and 4 Å, and later demonstrated a sharp rise to 7 Å with fluctuations in the order of 1–3 Å from 200 to 400 ns timepoint. However, from 400 to 500 ns, the protein RMSD achieved equilibrium between 6 and 8 Å RMSD (Fig. 4a). Changes in protein RMSD in the order of 1–3 Å are considered suitable for small proteins, indicating that the protein is not undergoing any significant conformational changes during the binding and simulation. Analysis of the reference ligand-6XPS complex RMSD graph, showed a steep rise in the protein RMSD from 2.5 to 7.2 Å in the first 50 ns of the simulation, and later fluctuated between 1.6 and 4.8 Å RMSDs till 400 ns duration. Post the 400 ns trajectory, RMSD stabilized at 2 Å. This indicates a slight conformational change in the protein (Fig. 4b). However, the overall RMSD average of both Violacein and reference ligand bound proteins are similar. The Violacein RMSD showed a major fluctuation at 100 ns trajectory reaching as high as 10 Å, and immediately stabilized between 4 and 6 Å. Again between 200 and 300 ns trajectory, the ligand RMSD was increased to 12–14 Å, however, from 300 to 500 ns trajectory, the Violacein ligand RMSD was found to achieve equilibrium between 4 and 6 Å. However, the reference (GDP) ligand RMSD, although stable for the initial 50 ns timeframe, was found to severely fluctuate between 8 and 56 Å through the entire duration of the simulation, averaging at 30 Å. Hence, the Violacein-ligand RMSD was lesser compared to the reference ligand. The graph shows the RMSD of the violacein ligand molecule more aligned on the RMSD of the protein. Overall, RMSD results indicate that the binding of the ligands at the active site of the 6XPS protein was stable and had little to no effect on the overall stability of the protein. The RMSF of the protein was calculated to characterize the local changes within the protein chain, and to calculate the flexibility of the backbone of protein after fitting to the frame of the reference during the simulation duration. Increased RMSF values indicate loosely structured loops and turns, whereas reduced flexibility signifies the presence of secondary structures such as helices and sheets. Cα atoms flexibility of each residue of the Violacein and reference-ligand protein complexes were determined. The graph’s peaks indicate the changes in the protein that fluctuate. Further, the contacts between the ligand and protein are represented with green lines. With the violacein-protein complex, the residues in the C terminal region were found to fluctuate reaching a high RMSF in the range of 9–10.5 Å and stabilized in the middle between 1.5 and 3 Å (Fig. 3c), while with the reference ligand-protein complex, residues in both the N and C terminal regions were increased and stabilized in the middle between 0.8 and 1.6 Å (Fig. 3d).

(a) Protein-Violacein RMSD (b) Protein-GDP RMSD (c) Violacein ligand RMSF (d) GDP ligand RMSF.
Before molecular docking, the secondary structure of the protein 6XPS was found to have major helices between the ranges 20–30, 45–50, 60–68, 82–90, 122–133, and 145–150, and beta sheets between the ranges 5–10, 33–40, 73–80, and 115–120, and the rest were looped regions (Fig. 5a). After subjecting to molecular docking with Violacein and subsequent molecular dynamics simulations the SSE was found to be made of major helices beta sheets. Slight fluctuations were observed at the C-terminal region (140–150), with some changes, while the other regions remained mostly unaffected (Fig. 5b). Similar secondary structure modifications were seen with the GDP-protein complex (Fig. 5c). Interestingly, the secondary structures of the protein during the 500ns simulation period (in complex with violacein) revealed 33.57% helices and 16.19% strands, making up 49.76% of the total secondary structure elements, while the protein during the 500 ns simulation period (in complex with GDP) was made up of 32.9% helices, 15.72% strands making up a total of 48.62% of the total secondary structures (Helices in bright red and strands in blue) (Fig. 5b, c).

(a) Protein Secondary Structure before molecular docking. (b) Protein Secondary Structure after docking with Violacein. (c) Protein Secondary Structure after docking with GDP ligand.
The Ligand RMSF indicative of the fluctuation within the ligand at atomic level detail and Violacein RMSF was observed to be stabilized between 3 and 4 Å, indicating few atomic fluctuations (Fig. 6a), while GDP ligand RMSF was found to be equilibrated over 20 Å values (Fig. 6b). GDP formed interactions with the residues GLU28, PHE59, and ASP110 for less than 30% of the total duration (Fig. 7a), while VAL149, VAL111, ASN114, LYS11, and GLY112 were found to form interactions with violacein for over 30% of the total simulation period (Fig. 7b). Amino acid residues LYS11, TYR51, VAL111, GLY112, ASN114, and VAL149 formed hydrogen bonds, LEU54, PHE59, LEU63, VAL111, and TYR150 formed hydrophobic bonds, and LYS11, TYR51, ARG104, VAL111, GLY112, ASN114, and HIS117 formed water bridges with violacein molecule (Fig. 7c).

(a) Ligand (violacein) RMSF. (b) Ligand (GDP) RMSF.

(a) GDP-6XPS contacts (b) Violacein-6XPS contacts (c) Histogram of Violacein-6XPS contacts occurring throughout the 500 ns simulation trajectory.
The radius of gyration (rGYr) represents the overall compactness of a protein during molecular simulation. The radius of gyration illustrates the moment of inertia of protein atoms relative to their centre of mass over a specific time interval. This was found mostly equilibrated at 4.6 Å, only one lowering to 4.2 Å in the violacein-protein complex, while in the GDP-protein complex, the rGYr varied between 4 and 4.8 Å. Overall, this indicated that the binding of the ligands to the 6XPS protein formed stable complexes. The Solvent Accessible Surface Area (SASA) of a protein is the surface area defined by its polar and non-polar interactions. The solvent-accessible surface area (SASA) of a protein diminishes as its compactness increases; thus, variations in SASA values can be utilized to assess protein stability. SASA values of violacein-protein complex (mostly stabilized between 160 and 240 Å), were lower than those of GDP-6XPS complex (300–600 Å). This indicated that the Violacein-6XPS complex was more compact and stable than the GDP-6XPS complex. Further, the ligand violacein was found to have no intramolecular hydrogen bonds throughout the simulation trajectory, although GDP molecule was found to have 1 intra-molecular bond. The calculated molecular surface area of violacein ligand with 1.4 Å probe radius was in the range of 304–308 Å2, while that of GDP ligand was higher (315–330 Å2). This is a measure equivalent to van der Waal’s surface area. The polar surface area, i.e., the total area exposed to solvent made of oxygen and nitrogen atoms ranged between 200 and 224 Å2 and 400–480 Å2 for the violacein and GDP molecules respectively (Fig. 8).

The ligand properties of the ligands during the simulation trajectory (a) Violacein ligand (b) GDP ligand.
MMGBSA and per residue analysis
The binding free energy of the ligand to the protein was quantitatively assessed using MMGBSA. The trajectory produced following the MD simulation was utilized for Thermal MM-GBSA. The molecular dynamics trajectories spanning 500 ns were utilized to estimate the binding free energy of both violacein and GDP with the 6XPS binding complexes, yielding 101 snapshots. The free energies for the Violacein-6XPS complex varied from − 31.00 to − 34.35 Kcal/mol, and from − 22.12 to − 32.75 Kcal/mol for GDP-6XPS complex (Fig. 9). The average binding energy for Violacein was − 31.89 ± 5.55 Kcal/mol, whereas the reference ligand exhibited − 26.62 ± 6.46 Kcal/mol. Further, the van der Waal’s energy, electrostatic solvation energy and total prime energies of these complexes were determined (Table 1). The results demonstrate the significant stability of both complexes and the consistent binding of the ligand at the active site. Overall, snapshots obtained at various intervals during the MD simulation demonstrated that the binding of violacein was more stable than that of GDP.
The contributions of individual amino acid residues involved in the binding interactions were quantified. The per residue interaction profile of VAL111, ASN114, LYS11, TYR51, ARG104, and PHE59 was analyzed. The binding energies attributed to these amino acid residues were − 5.19, − 4.5, − 6.03, − 5.63, − 9.14, and − 8.3 Kcal/mol, respectively and were lower compared to those by GDP (Table 2). Further, it was confirmed that the molecules were bound at the active site of the protein. The crucial role of these residues in complex formation was emphasized.

MM-GBSA binding free energy values throughout the 500ns molecular dynamics simulation (101 snapshots of the trajectory).
Conceptual DFT calculation
Chemical reactivity, charge transfer, and thermodynamic and kinetic stability of the molecule are all significantly influenced by the energy band gap i.e., the difference in energies between the highest and lowest occupied molecular orbitals (HOMO and LUMO respectively)43. The HOMO and LUMO energies also define the ability of a compound to either receive or donate electrons. These also help with the prediction of the most reactive conformations of the compound. Small energy gaps make molecules more polarizable, causing them to be soft molecules with high chemical reactivity and low kinetic stability. On the other hand, molecules with a wider energy gap provide additional kinetic steadiness and a reduced amount of chemical reactivity44. Figures 10 and 11 illustrate the energies of HOMO and LUMO, and the electrostatic potential surface of Violacein and the reference ligand (GDP), respectively. Energies of HOMO and LUMO for the Violacein ligand were calculated to be − 0.031818 eV and − 0.040749 eV, respectively, while those for the reference ligand were − 0.017180 eV and − 0.132092 eV, respectively. Consequently, the energy band gap (ΔEHOMO-ELUMO) for the violacein molecule was calculated to be − 0.072567 eV, whereas for the reference ligand, it was found to be 0.149272 (Table 3). These findings suggest that violacein, with the smaller energy gap, is more polarizable with higher chemical reactivity. The electronegativity, chemical hardness and softness, net electrophilicity index, and chemical potential of the violacein molecule were also identified as additional quantum mechanical reactivity descriptors using the HOMO and LUMO energies (Table 3). The electrophilicity index provides information regarding the binding ability of a compound with other biomolecules and measures the equilibrium between a particle’s resistance to changing its electron density in response to its surroundings and its inclination to attract additional electron density45. Electrostatic potential maps for compounds Violacein and GDP revealed regions of positive and negative charge, key for understanding their binding interactions. The molecular electrostatic potential surface will illustrate the three-dimensional charge distributions of molecules. The molecular electrostatic potential surface serves as a representation of regions with electron excess and deficiency46. The electrostatic potential of Violacein and the reference ligand were − 120,979 and − 153.785 which highlights the areas where these compounds might attract electrophiles or nucleophiles. Violacein, with a much more negative potential, is likely to have stronger interactions with positively charged species (electrophiles), indicating a higher capacity for engaging in such interactions compared to GDP, which has a much smaller negative potential. This implies that Violacein may be more reactive and can form stronger bonds in biological systems or during drug interactions, making it potentially a more effective compound for targeting positively charged biomolecules.

3D plot of HOMO, LUMO and molecular electrostatic potential surface of violacein compound.

3D plot of HOMO, LUMO and molecular electrostatic potential surface of GDP molecule.
Discussion
The United Nations ad hoc interagency coordinating group on antimicrobial resistance cautioned that drug-resistant infections to be responsible for up to 10 million fatalities per annum by 2050 and cause economic harm. Antimicrobial resistance may become responsible for extreme poverty in nearly 24 million of the population by 2030 47. Owing to the intrinsic limits of currently available antifungal medications and the growth in antifungal resistance, treatment often necessitates intensive antifungal therapy. Hence, there is an increased attention towards studying crucial metabolic pathways to comprehend the intricacies of fungal biology in the background of human disease and find possible targets for antifungals. One of these is the process of the fungal de novo purine biosynthesis pathway. The purine biosynthesis and salvage pathways of several clinically significant pathogenic fungi, such as Candida albicans, A. fumigatus, and Cryptococcus neoformans, have been thoroughly explored48. Serious virulence reduction or, in some cases, complete loss of virulence is related to genetically disrupting the purine biosynthesis process49. To lower the likelihood of infection while simultaneously considering antifungal resistance, the focus has now shifted to identifying novel drug targets across unconventional pathways occurring across fungal biosystems. One possible target identified is the protein nucleoside diphosphate kinase (Ndk), which is significant for the nucleotide biosynthesis of organisms50.
The reversible exchange of the γ-phosphate between nucleoside triphosphate and nucleoside diphosphate is catalysed by the enzyme Ndk, and maintains intracellular nucleotide triphosphates, which are required for DNA and RNA synthesis50,51. Recent studies identified Ndk to play an elaborate role in the development of spores and sclerotia in Aspergillus species and, hence, may be considered a probable antifungal drug target52,53. The crucial functions performed by Ndk in the survival of fungi and their virulence increases the enzyme’s potential as a novel antifungal target50. The primary role of the highly conserved enzyme Ndk, which is found worldwide, is to maintain intracellular NTPs. The ultimate step of the purine and pyrimidine biosynthesis pathway is catalysed by the enzyme Ndk by converting nucleoside diphosphates into nucleoside triphosphates and producing ATP54. These fundamental components serve as signal transduction pathway mediators and are necessary for the production of DNA and RNA. For fungi to survive in the natural environment, controlled synthesis of NTPs is essential. Particularly in Eumycophyta, a range of biological roles for NDK have gained insufficient attention. A. fumigatus must adjust to variations in nucleoside accessibility to survive and flourish55,56. Ndk is a key enzyme in the final step of nucleotide biosynthesis, which is essential for fungal survival. Inhibition of Ndk, would cause the inhibition of DNA and RNA synthesis, it would lead to a wide variety of effects within the fungus, including cessation of growth, reproduction as well as cell death50,56,57. Hence, since Ndk aids in survival inside human hosts, inhibition of the NDK protein may help fight against A. fumigatus. Hence, Ndk has been discovered as a vital gene in both A. fumigatus and A. nidulans, suggesting it may be a good target for new antifungals58. Ndk has also been investigated as a probable target for creating antiparasitic medications in parasitic infections such as Leishmaniasis and trypanosomiasis59. Similarly, research has been focused on Aspergillus flavus Ndk for antifungal treatments60. In line with its critical functions in DNA synthesis, A. nidulans Ndk has been linked to vital cell cycle events such as hyphal expansion. In contrast, Ndk of A. flavus has been observed to play a complex function in the development of spores, conidia, and sclerotia and has been demonstrated to contribute to plant pathogenicity52,56,60. The rise in drug-resistant fungi has prompted the exploration of natural compounds as possible antifungal agents. Hence, the present work attempted to evaluate the efficacy of violacein, a natural bacterial pigment, for its anti-fungal effects, specifically on A. fumigatus.
Bioinformatics uses various computation techniques to generate hypotheses or uncover novel biological phenomena, such as sequence and structural alignment, and investigation of a sizable assortment of biological data. Gaining pace is using bioinformatics tools and methods to analyse the growing volume of data produced in genomics, molecular biology, transcriptomics, and proteomics61. Additionally, the expanse of data compiled in databases and literature for creating molecular profiles and gathering information on the epidemiology of pathogens has significantly expanded35,62. Therefore, it is essential to apply computational techniques and approaches to avert the spread of drug-resistant microbes, detect and classify pathogens, find indicators for diagnosis and treatment, enable individualised interventions, and forecast patient outcomes. The interactions with the target (Ndk) were analysed using bioinformatic tools. The STRING analysis depicted the interactions with 11 nodes and 49 edges. Many of the identified enzymes are involved in phosphorylation reactions, essential for conversion of nucleoside monophosphates to diphosphates. Thymidylate kinase, uridylate kinase, and guanylate kinase facilitate the transformation of their corresponding monophosphates into diphosphates. Ndk subsequently transforms these NDPs into NTPs, which are essential for DNA and RNA synthesis63,64,65. Adk1 and Adk2 regulate the ADP/ATP equilibrium65,66,67. Cytidine triphosphate synthase catalyzes the conversion of uridine triphosphate to cytidine triphosphate68,69,70,71. Similarly, the other identified enzyme, Similarly, the other identified enzyme, Ribonucleoside-diphosphate reductase is responsible for ribonucleotides to deoxyribonucleotides, which are required for the action of NDK40. Hence, all these proteins are essential for microorganism functioning, including Ndk.
Hence, this study aimed at the evaluation of violacein as an antifungal against the Nucleoside Diphosphate Kinase protein of A. fumigatus, via computational methods. Investigating and analysing the Violacein-Ndk protein interactions in silico revealed that the molecule significantly interacts with the majority of the active amino acid residues of both enzymes’ catalytic pockets with robust interactions, primarily H-bonds, hydrophobic interactions, ionic bonds, and water bridges, of relatively short bond distances and low binding energies. Interaction analysis of violacein with Ndk revealed four hydrogen bonds with the positively charged amino acid residues ARG87, THR93, ARG104, and ASN114, two salt bridges with the positively charged residues LYS11 and ARG104, and two Pi-Pi Stalking interactions [with the hydrophobic amino acid residue PHE59]. A glide g-score of − 7.211 kcal/mol was observed for the same interaction. In comparison, the chosen reference ligand (GDP, pre-bound to 6XPS) was found to generate a complex with glide g-score of − 7.010 Kcal/mol, forming 4 hydrogen bonds (ASN114, THR93, HIE117, GLY118), and 2 Pi-Pi stalking interactions (PHE59). The lower glide gscore of GDP-Ndk complex (− 7.01 Kcal/mol), and lower number of total interactions within when compared to Violacein-Ndk complex (− 7.211 Kcal/mol), indicate a stronger binding of violacein to the protein.
The study analyzed the dynamics variation in the Cα backbone of both the generated complexes. The calculated protein overall RMSD average for both violacein and reference ligand was comparable, however, violacein-protein complex had a lower RMSD value. The protein RMSF value in both complexes was analyzed and were found to be fairly stable expect at the N and C terminal regions. The secondary structures of the protein (in complex with violacein) revealed 33.57% helices and 16.19% strands, making up 49.76% of the total secondary structure elements, and comparison with pre-bound protein revealed minimal changes to the protein’s secondary structural elements. Comparison of the ligand properties, the ligand RMSF indicated fluctuation within the ligand at atomic level detail, with Violacein RMSF stabilized between 3 and 4 Å, while GDP ligand RMSF was found to be equilibrated over 20 Å values. Violacein-Ndk complex was found to be more compacted and stable than the reference ligand-protein complex. VAL149, VAL111, ASN114, LYS11, and GLY112 were found to form interactions with violacein for over 30% of the total simulation period and GLU28, PHE59, and ASP110 for less than 30% of the total duration with GDP. Further MMGBSA analysis of the 101 snapshots of the trajectory revealed an energy of − 31.89 ± 5.55 Kcal/mol for the violacein complex, which was lower than that of the GDP complex. In addition, the study quantified the contributions of individual amino acid residues in binding interactions, analyzing the per residue interaction profiles of VAL111, ASN114, LYS11, TYR51, ARG104, and PHE59. The binding energies were lower than those by GDP, indicating that the molecules were bound at the active site of the protein. The crucial role of these residues in complex formation was emphasized. The Violacein-Ndk complex exhibited superior stability in both protein and ligand RMSD analyses when compared to the GDP-Ndk complex. The Violacein-Ndk complex exhibited enhanced binding interactions, a more compact structure, and increased stability as indicated by the MMGBSA binding energy analysis. This suggests that Violacein establishes a more stable and consistent interaction with the NDK protein compared to the reference ligand, thereby positioning it as a more effective ligand for protein binding.
The DFT studies of Violacein, particularly its HOMO and LUMO energies, offer essential insights into its chemical reactivity and stability. Compared to the reference ligand (ΔE = 0.114917 eV), the violacein molecule was calculated to have a smaller energy band gap (ΔE = 0.072567 eV), suggesting its softness with lower kinetic stability and higher chemical reactivity. This minor gap increases the polarizability of violacein, facilitating its participation in electron transfer during biochemical interactions. The electrostatic potential analysis corroborates this, indicating that Violacein exhibits a substantially more negative potential (− 120979) compared to the reference ligand (− 153.785), suggesting enhanced interactions with electrophiles and positively charged biomolecules. These attributes indicate the possible effectiveness of Violacein as an antifungal agent, particularly through its interactions with fungal proteins such as the Ndk protein.
These DFT calculations were performed using the Becke 3-parameter exchange functional (B3LYP) method with the 6-31G(d, p) basis set, which has both merits and limitations. The primary advantage of using the 6-31G(d, p) basis set is its incorporation of polarization functions (d on heavy atoms and p on hydrogen), which improves the description of electron distribution and molecular geometry, especially in the regions of electron density critical for bonding. This results in enhanced accuracy in predicting molecular properties, including dipole moments, and reaction mechanisms, rendering it a favoured option for organic compounds such as violacein. However, the 6-31G(d, p) basis set is not without limitations. It may not consistently yield highly accurate results for systems characterized by substantial electron correlation effects or extensively delocalized electrons, due to its medium-sized basis set, as for larger or more complex molecules, it may be inadequate relative to larger basis sets or more advanced methods that successfully address electron correlation and dispersion interactions. Furthermore, more refined datasets (such as 6-311 + + G**) could yield improved precision at the expense of more computational resources, yet it provides a reasonable trade-off between accuracy and computational efficiency. Thus, while 6-31G(d, p) offers solid performance for Violacein, its limitations must be considered, particularly when higher accuracy is required.
There exists no direct evidence which ascertains the antifungal activity of Violacein. Notwithstanding, damages in the fungal cell wall, decreased hyphal development and biofilm formation owing to induction of ROS by violacein is a strongly profounded hypothetical mechanism(s). NDK was chosen as the potential target for this study due to its importance in fungal growth and development as established72 based on the extensive review of literature and in silico screening, by molecular docking conducted by us, we did not come across any other significant pathway through which violacein could show the antifungal activity against A. fumigatus. In addition, the existing non-NDK-antifungal drugs are reported for their adverse effects73.
This suggests that Violacein could be an effective molecule and strong antifungal agent against the NDK protein of A. fumigatus. Violacein’s potential as an antifungal against A. fumigatus was discovered through in silico techniques via inhibition of the Nucleoside Diphosphate Kinase protein. The study provides a strong platform for future evaluation of the pigment as an anti-fungal agent in vitro and in vivo, suggesting the pigment as a potential candidate in the management of A. fumigatus infections.
Materials and methods
STRING analysis
STRING analysis was carried out to look for known and predicted interactions between proteins. Version 11.5 of the STRING application (https://string-db.org/) was used74. The protein was submitted to the database and performed with STRING’s default parameters. A full STRING network with a minimum required interaction score of 0.4 was employed, the network edges were evidence-based, and active interaction sources were based on neighborhood, experiments, text mining, gene fusion, databases, co-occurrence, and co-expression.
Protein preparation
The NDK (PDB ID: 6XPS) X-ray crystal structures of the protein was chosen and obtained from the RCSB PDB with a resolution of 1.64 Å and chain A of sequence length 162. The protein was then included in the Maestro workspace (V 14.1)72,75. The protein was loaded into the protein preparation wizard, and the plan for minimizing protein energy was implemented. The pH was set to 7.5, and the Prime module was used to fill in the missing chains and loops. The protein’s regulatory component was removed, another application of the refining process. The Epik module was used to assign the appropriate protonation state and hydrogen atoms were added at pH 7.5. Optimization of the protein was achieved using PROPKA pH 7.5, the OPLS4 force field was used to minimize atoms beyond 0.3 Å RMSD, and water molecules at a distance of over 5 Å were also removed, and partial atomic charges were assigned. The reduced protein was then further processed for grid construction in the receptor grid-generating panel to reflect the active binding pocket of the protein to the ligand for Glide docking. The pre-existing ligand (GDP) in the selected protein was chosen to carry out the construction of grid. Receptor grid creation was then performed with the default settings, scaling the van der Waals radius, scaling factor 1.0, and removing the partial charge. The residues around the pre-bound ligand were identified to be LYS11, TYR51, LEU54, PHE59, LEU63, TYR66, ARG87, GLY91, ALA92, THR93, ARG104, VAL111, GLY112, ARG113, ASN114, CYS116, HIS117, GLY119, ASP120.
Ligand preparation
The structure of violacein was taken from the PubChem database (PubChem CID:11053) to optimize energy and produce a precise 3D structure. The ligand preparation step was essential to molecular docking, generating optimum structures suitable for molecular docking and eliminating ligand errors. Using the Epik classic module, with OPLS4 forcefield, energy minimization was then carried out while preserving a pH of 7 in the ionization state, defining desalt, single tautomer synthesis, and maintaining specified chirality.
Molecular docking
In the Maestro interface of the Schrödinger suite, Glide is a very efficient way of detecting ligand hits and aids in lead optimization in structure-based virtual screening via molecular docking. Molecular docking is a technique that evaluates how tiny molecules behave in the binding pocket of the target protein and reveals important biological processes while keeping the receptor rigid72,76,77,78. The chosen protein was subjected to molecular docking with violacein using Glide XP (enhanced precision) docking. Validation of the docking protocol was done with the superposition of pre-bound reference molecule with itself post-docking79,80,81. The optimum docking pose was chosen based on additional examination of the glide g-score and molecular interactions with the protein. The chosen complex was further subjected to MM-GBSA to minimize and refine the protein-ligand complex. This step was performed in the VGSB solvation model, and OPLS4 forcefield was applied.
Molecular dynamics simulation
The functions and dynamics of protein-ligand complexes have long been studied using the DESMOND v. 13.7 System Builder workflow. Desmond MD simulations of the non-bonding interaction between the ligand and the protein were done for 500 ns for the protein 6XPS to understand the stability of the violacein-6XPS complex better. The entire system was submerged in a TIP3P water-solvent model. The orthorhombic box of size 10 × 10 × 10 Å of boundary condition was maintained throughout the system development process. The OPLS4 force field was prepared using the System Builder program and further neutralized by adding 0.15 M NaCl to the buffer. Additionally, minimization was accomplished using the minimizing tool. The violacein-6XPS developed system consisted of 21,177 atoms in total and that of GDP-6XPS had 21,210 atoms. The MD process’s temperature (K) and pressure (bar) were held constant throughout the experiment at 300 K and 1.01325 bar, respectively, and the calculation was performed for 500 ns. Subsequently, the simulated interaction diagram tool built with Desmond generated reports82,83.
Post-dynamics analysis
The MMGBSA was executed utilizing a script to compute the binding free energy of the Violacein-6XPS complex and the reference ligand-6XPS complex. Every tenth frame of 1000 frames produced from molecular dynamics simulation was utilized to determine binding energy. Employing the OPLS4 force field and the VSGB 2.1 solvent model, the binding energies of ligands were computed using the formula provided below.
Ecomplex represents the total free energy of the protein–ligand complex, while Eligand and Ereceptor denote the free energies of the ligand and receptor in a solvent, respectively.
Per-residue decomposition was conducted to quantitatively analyze the energetic contributions of each amino acid, encompassing both backbone and sidechain assessments. The study of binding energy decomposition was essential for outlining the significance made by every residue to the ligand.
Conceptual density-functional theory (DFT) calculation
DFT-based calculations aim to analyze the interaction of a compound with a receptor protein. DFT calculation for violacein and GDP molecules were analysed using optimization module of the Jaguar v11.7 package. To study the reactivity of Violacein and GDP reference molecule, the DFT calculations focused on geometry optimization, employing the B3LYP functional combined with the 6-31G(d, p) basis set, primarily focusing on the highest and lowest occupied molecular orbitals (HOMO and LUMO, respectively). Electron density, electrostatic potential, surface, spin density, and molecular orbitals parameters were selected. Additionally, the band energy gap was calculated as the difference in energies between HOMO and LUMO to assess electronic properties84. This approach allows for a comprehensive investigation of Violacein and GDP molecule reactivity and potential interactions with the receptor protein.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Seagle, E. E., Williams, S. L. & Chiller, T. M. Recent trends in the epidemiology of fungal infections. Infect. Dis. Clin. North. Am. 35, 237–260 (2021).
Brown, G. D. et al. Hidden killers: human fungal infections. Sci. Transl Med. 4, 165rv13 (2012).
Fisher, M. C., Hawkins, N. J., Sanglard, D. & Gurr, S. J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Sci. (1979). 360, 739–742 (2018).
Verweij, P. E. et al. The one health problem of azole resistance in Aspergillus fumigatus: current insights and future research agenda. Fungal Biol. Rev. 34, 202–214 (2020).
Rhodes, J. & Fisher, M. C. Global epidemiology of emerging Candida Auris. Curr. Opin. Microbiol. 52, 84–89 (2019).
Arastehfar, A. et al. Drug-resistant Fungi: an emerging challenge threatening our limited antifungal armamentarium. Antibiot. (Basel) 9 (2020).
Doughty, K. J., Sierotzki, H., Semar, M. & Goertz, A. Selection and amplification of fungicide resistance in Aspergillus fumigatus in relation to DMI fungicide use in agronomic settings: hotspots versus coldspots. Microorganisms 9, (2021).
Latgé, J. P. & Chamilos, G. Aspergillus Fumigatus and aspergillosis in 2019. Clin. Microbiol. Rev. 33, (2019).
Pugliese, M., Matić, S., Prethi, S., Gisi, U. & Gullino, M. L. Molecular characterization and sensitivity to demethylation inhibitor fungicides of aspergillus fumigatus from orange-based compost. PLoS One. 13, e0200569 (2018).
Lass-Flörl, C. How to make a fast diagnosis in invasive aspergillosis. Med. Mycol. 57, S155–S160 (2019).
Liu, D. Molecular Detection of Human Bacterial Pathogens (CRC, 2011).
Paterson, R. R. M. & Lima, N. Molecular Biology of Food and Water Borne Mycotoxigenic and Mycotic Fungi (CRC, 2015).
Brown, G. D. et al. Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165 (2012).
Ariza-Heredia, E. J. & Kontoyiannis, D. P. Our recommendations for avoiding exposure to fungi outside the hospital for patients with haematological cancers. Mycoses 57, 336–341 (2014).
Marr, K. A. et al. Fungal infection prevention after hematopoietic cell transplantation. Bone Marrow Transpl. 44, 483–487 (2009).
Hoenigl, M. & Krause, R. Antifungal therapy of aspergillosis of the central nervous system and aspergillus endophthalmitis. Curr. Pharm. Des. 19, 3648–3668 (2013).
Paterson, R. R. M. & Lima, N. Filamentous fungal human pathogens from food emphasising Aspergillus, Fusarium and Mucor. Microorganisms 5, (2017).
CDC. Antibiotic resistance threats in the United States, (2019). www.cdc.gov/DrugResistance/Biggest-Threats.html (2019).
Kakde, U. & Kakde, H. Incidence of post-harvest disease and airborne fungal spores in a vegetable market. Acta Bot. Croat 71, (2012).
Torimiro, N., Makinde, I., Omole, R. & Daramola, O. Deterioration profile of postharvest onion (Allium cepa L.) bulbs Induced by potential pathogenic microorganisms. Int. J. Pathogen Res. 5, 39–45 (2020).
Vicente, M. F., Basilio, A., Cabello, A. & Peláez, F. Microbial natural products as a source of antifungals. Clin. Microbiol. Infect. 9, 15–32 (2003).
Tripathi, R. K. & Gottlieb, D. Mechanism of action of the antifungal antibiotic pyrrolnitrin. J. Bacteriol. 100, 310–318 (1969).
Elkhayat, E. S. & Goda, A. M. Antifungal and cytotoxic constituents from the endophytic fungus Penicillium Sp. Bull. Fac. Pharm. Cairo Univ. 55, 85–89 (2017).
Tamehiro, N. et al. Bacilysocin, a novel phospholipid antibiotic produced by Bacillus subtilis 168. Antimicrob. Agents Chemother. 46, 315–320 (2002).
John Jimtha, C., Jishma, P., Sreelekha, S., Chithra, S. & Radhakrishnan, E. K. Antifungal properties of prodigiosin producing rhizospheric Serratia sp. Rhizosphere 3, 105–108 (2017).
Mahmoud, S., Ziedan, E. S., Farrag, E., Khalaphallah, R. & Elymani, A. Antifungal activity of pyocyanin produced by pseudomonas aeruginosa against fusarium oxysporum schlech a root-rot phytopathogenic fungi. 9, 43–50 (2016).
Dawoud, T. et al. Characterization and antifungal activity of the yellow pigment produced by a Bacillussp. DBS4 isolated from the lichenDirinaria agealita. Saudi J. Biol. Sci. 27, (2019).
Dike-Ndudim, J., Ugenyi, L. & Ndubueze, C. Assessment of antifungal potentials of violacein extract from Chromobacterium violaceum isolated from domestic and recreational water sources in Owerri, Imo State, Nigeria. World J. Adv. Res. Rev. 10, 168–172 (2021).
Durán, N. et al. Violacein and its antifungal activity: comments and potentialities. Lett. Appl. Microbiol. 75, 796–803 (2022).
Dogancı, M. A. et al. Investigation of potential inhibitor properties of violacein against HIV-1 RT and CoV-2 spike RBD:ACE-2. World J. Microbiol. Biotechnol. 38, 161 (2022).
Sasidharan, A. et al. Antifungal activity of violacein purified from a novel strain of Chromobacterium sp. NIIST (MTCC 5522). J. Microbiol. 53, 694–701 (2015).
Becker, M. H., Brucker, R. M., Schwantes, C. R., Harris, R. N. & Minbiole, K. P. C. The bacterially produced metabolite violacein is associated with survival of amphibians infected with a lethal fungus. Appl. Environ. Microbiol. 75, 6635–6638 (2009).
Sen, T., Barrow, C. J. & Deshmukh, S. K. Microbial Pigments in the food industry—challenges and the way forward. Front. Nutr. 6 (2019).
Venil, C. K. et al. Spray drying of violet pigment from Chromobacterium violaceum UTM 5 and its application in food model systems. Int. Biodeterior. Biodegrad.. 102, 324–329 (2015).
Carriço, J. A., Sabat, A. J., Friedrich, A. W. & Ramirez, M. Bioinformatics in bacterial molecular epidemiology and public health: databases, tools and the next-generation sequencing revolution. Euro. Surveill. 18, 20382 (2013).
Müller-Dieckmann, H. J. & Schulz, G. E. The structure of Uridylate kinase with its substrates, showing the transition state geometry. J. Mol. Biol. 236, 361–367 (1994).
Narvaez-Ortiz, H. Y., Lopez, A. J., Gupta, N. & Zimmermann, B. H. A CTP synthase undergoing stage-specific spatial expression is essential for the survival of the intracellular parasite Toxoplasma Gondii. Front. Cell. Infect. Microbiol. 8, 83 (2018).
Alam, M. S., Moriyama, H. & Matsumoto, M. Enzyme kinetics of dUTPase from the planarian Dugesia ryukyuensis. BMC Res. Notes. 12, 163 (2019).
Li, Y., Zhang, Y. & Yan, H. Kinetic and thermodynamic characterizations of yeast guanylate kinase. J. Biol. Chem. 271, 28038–28044 (1996).
Torrents, E. Ribonucleotide reductases: essential enzymes for bacterial life. Front. Cell. Infect. Microbiol. 4, 52 (2014).
Klepinin, A. et al. Adenylate kinase and metabolic signaling in cancer cells. Front. Oncol. 10, Preprintat (2020).
Van Rompay, A. R., Johansson, M. & Karlsson, A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol. Ther. 87, 189–198 (2000).
Velmurugan, G., Vedha, S. A. & Venuvanalingam, P. Computational evaluation of optoelectronic and photophysical properties of unsymmetrical distyrylbiphenyls. RSC Adv. 4, 53060–53071 (2014).
Thanikaivelan, P., Subramanian, V., Rao, J. R. & Nair, B. U. Application of quantum chemical descriptor in quantitative structure activity and structure property relationship. Chem. Phys. Lett. 323, 59–70 (2000).
Domingo, L. R., Ríos-Gutiérrez, M. & Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21, (2016).
Roohzadeh, R. & Mahdavi, M. Prediction of explosive properties of newly synthesized amino nitroguanidine-based energetic complexes via density functional theory. J. Mol. Model. 26, 1–11 (2020).
New report calls for urgent action to avert antimicrobial resistance crisis. https://www.who.int/news/item/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis (2019).
Chitty, J. L. et al. GMP synthase is required for virulence factor production and infection by Cryptococcus neoformans*. J. Biol. Chem. 292, 3049–3059 (2017).
Chitty, J. L. & Fraser, J. A. Purine acquisition and synthesis by human fungal pathogens. Microorganisms 5, (2017).
Nguyen, S., Jovcevski, B., Pukala, T. L. & Bruning, J. B. Nucleoside selectivity of aspergillus fumigatus nucleoside-diphosphate kinase. FEBS J. 288, 2398–2417 (2021).
Monk, B. C. et al. Fungal lanosterol 14α-demethylase: a target for next-generation antifungal design. Biochim. Biophys. Acta BBA Proteins Proteom. 1868, 140206 (2020).
Wang, Y. et al. Molecular and structural basis of nucleoside diphosphate kinase–mediated regulation of spore and sclerotia development in the fungus aspergillus flavus. J. Biol. Chem. 294, (2019). jbc.RA119.007505.
Nguyen, S., Jovcevski, B., Pukala, T. L. & Bruning, J. B. Structural insights into the antifungal drug target guanosine monophosphate synthase from Aspergillus Fumigatus. Acta Crystallogr. D Struct. Biol. 78, 248–259 (2022).
Chakrabarty, A. M. Nucleoside diphosphate kinase: role in bacterial growth, virulence, cell signalling and polysaccharide synthesis. Mol. Microbiol. 28, 875–882 (1998).
Dinamarco, T. M. et al. Aspergillus Fumigatus calcineurin interacts with a nucleoside diphosphate kinase. Microbes Infect. 14, 922–929 (2012).
Lin, X., Momany, C. & Momany, M. SwoHp, a nucleoside diphosphate kinase, is essential in aspergillus nidulans. Eukaryot. Cell. 2, 1169–1177 (2003).
Wang, Y. et al. Molecular and structural basis of nucleoside diphosphate kinase-mediated regulation of spore and sclerotia development in the fungus aspergillus flavus. J. Biol. Chem. 294, 12415–12431 (2019).
Dinamarco, M. Aspergillus Fumigatus calcineurin interacts with a nucleoside diphosphate kinase. Microbes Infect. 14, 922–929 (2012).
Souza, T. A. C. B. et al. Molecular adaptability of nucleoside diphosphate kinase b from trypanosomatid parasites: stability, oligomerization and structural determinants of nucleotide binding. Mol. Biosyst. 7, 2189–2195 (2011).
Wang, Y. et al. Molecular and structural basis of nucleoside diphosphate kinase–mediated regulation of spore and sclerotia development in the fungus aspergillus flavus. J. Biol. Chem. 293, 12415–12431 (2019).
Hendriksen, R. S. et al. Using genomics to track global antimicrobial resistance. Front. Public. Health. 7, 242 (2019).
Bhat, S. S., R, S. & Prasad, S. K. A Bioinformatics approach towards plant-based anticancer drug discovery. In Computational Approaches in Biotechnology and Bioinformatics (eds Pathak, D. P., Raut, R., Jaramillo-Isaza, S., Borkar, P. & Jhaveri, R. H.) (CRC, 2024).
Cai, X. et al. The atypical guanylate kinase MoGuk2 plays important roles in Asexual/Sexual development, conidial septation, and pathogenicity in the Rice Blast Fungus. Front. Microbiol. 8, (2017).
Cui, Q. et al. Thymidylate kinase: an old topic brings new perspectives. Curr. Med. Chem. 20, 1286–1305 (2013).
Wang, L. The role of Ureaplasma nucleoside monophosphate kinases in the synthesis of nucleoside triphosphates. FEBS J. 274, 1983–1990 (2007).
Claypool, S. M., Oktay, Y., Boontheung, P., Loo, J. A. & Koehler, C. M. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell. Biol. 182, 937–950 (2008).
Miura, K. et al. Cloning and characterization of adenylate kinase fromChlamydia pneumoniae. J. Biol. Chem. 276, 13490–13498 (2001).
Endrizzi, J. A., Kim, H., Anderson, P. M. & Baldwin, E. P. Crystal structure of Escherichia coli cytidine triphosphate synthetase, a nucleotide-regulated glutamine amidotransferase/ATP-dependent amidoligase fusion protein and homologue of anticancer and antiparasitic drug targets. Biochemistry 43, 6447–6463 (2004).
Lieberman, I. Enzymatic amination of uridine triphosphate to cytidine triphosphate. J. Biol. Chem. 222, 765–775 (1956).
Chakraborty, K. P. & Hurlbert, R. B. Role of glutamine in the biosynthesis of cytidine nucleotides in Escherichia coli . Biochim. Biophys. Acta. 47, 607–609 (1961).
Long, C. W. & Pardee, A. B. Cytidine triphosphate synthetase of Escherichia coli B: I. Purification and kinetics. J. Biol. Chem. 242, 4715–4721 (1967).
Prasad, S. K. et al. Naringin from coffee inhibits foodborne aspergillus fumigatus via the NDK pathway: evidence from an in silico study. Molecules 28, (2023).
Low, Y. et al. Triazolopyrimidine herbicides are potent inhibitors of aspergillus fumigatus acetohydroxyacid synthase and potential antifungal drug leads. Sci. Rep. 11 (2021).
STRING Analysis. https://string-db.org/
Schrödinger Release 2024-3, Glide, Schrödinger.
Bhat, S. S., Mahapatra, S., Das, Sommano, S. R. & Prasad, S. K. Virtual screening and quantitative structure–activity relationship of Moringa oleifera with Melanoma Antigen A (MAGE-A) genes against the therapeutics of Non-small Cell Lung cancers (NSCLCs). Cancers (Basel). 14, 5052 (2022).
Bhat, S. S. et al. In silico examination of peptides containing selenium and ebselen backbone to assess their Tumoricidal potential. Silico examination of peptides containing selenium and Ebselen Backbone to assess their Tumoricidal potential. https://doi.org/10.55691/2278-344X.1029 (2021).
Bhat, S. S. & Prasad, S. K. In silico screening of violacein as an epidermal growth factor receptor inhibitor. Int. J. Health Allied Sci. 11, 6 (2022).
Dalal, V., Golemi-Kotra, D. & Kumar, P. Quantum mechanics/molecular mechanics studies on the catalytic mechanism of a novel esterase (FmtA) of Staphylococcus aureus. J. Chem. Inf. Model. 62 (2022).
Kumari, R., Rathi, R., Pathak, S. & Dalal, V. Structural-based virtual screening and identification of novel potent antimicrobial compounds against YsxC of Staphylococcus aureus. J. Mol. Struct. 1255, 132476 (2022).
Kumari, R. & Dalal, V. Identification of potential inhibitors for LLM of Staphylococcus aureus: structure-based pharmacophore modeling, molecular dynamics, and binding free energy studies. J. Biomol. Struct. Dyn. 40, 1–15 (2021).
Jahagirdar, S. et al. In silico evaluations of phytochemicals from Withania somnifera exhibiting anticancer activity against NAD[P]H-quinone oxidoreductase. Hum. Exp. Toxicol. 43, 09603271241291399 (2024).
Research, D. E. S. Schrödinger Release 2024-3: Desmond Molecular Dynamics System.
Bhat, S. S. et al. Preliminary evaluation of lablab purpureus phytochemicals for anti-BoHV-1 activity using in vitro and in silico approaches. ACS Omega. https://doi.org/10.1021/acsomega.3c01478 (2023).
Acknowledgements
S.R, S.S.B, C.D, C.S, B.S, D.D and S.K.P thank JSS Academy of Higher Education and Research (JSS AHER), Mysuru, Karnataka, India, for the infrastructural support necessary for conduct of this research. J.S and S.R.S thank Chiang Mai University (CMU), Thailand, for supporting this research. S.P.K acknowledge and thank Amrita Vishwa Vidyapeetham for the infrastructure and support provided. The authors acknowledge and extend their appreciation to the Researchers Supporting Project Number (RSPD2024R748), King Saud University, Riyadh, Saudi Arabia for supporting this study.
Funding
This research was funded by King Saud University, Riyadh, Saudi Arabia, project number (RSPD2024R748).
Author information
Authors and Affiliations
Contributions
S.R: Conceptualization, Supervision, Writing - original draft, Writing- review & editing; S.S.B: Methodology, Writing - original draft, Writing- review & editing, Formal analysis, Investigation, Visualization; J.S: Supervision, Writing - original draft, Formal analysis, Investigation; C.D: Methodology, Writing- review & editing, Formal analysis, Investigation; B.S: Writing- review & editing, Data curation, Visualization; C.S: Supervision, Writing- review & editing, Resources, Project administration; D.D: Supervision, Writing- review & editing, Resources, Data curation, Project administration; S.F.A: Writing- review & editing, Formal analysis, Funding acquisition, Resources; S.M.A: Writing- review & editing, Formal analysis, Funding acquisition, Resources; S.P.K: Writing - original draft, Writing- review & editing, Formal analysis, Validation; S.R.S: Conceptualization, Supervision, Writing - original draft, Formal analysis, Investigation; S.K.P: Conceptualization, Supervision, Funding acquisition, Resources, Validation, Software, Project administration.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sindhu, R., Bhat, S.S., Sangta, J. et al. Gaining molecular insights towards inhibition of foodborne fungi Aspergillus fumigatus by a food colourant violacein via computational approach. Sci Rep 14, 29905 (2024). https://doi.org/10.1038/s41598-024-81471-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-81471-2
