
Abstract
Patients with metabolic dysfunction-associated steatotic liver disease (MASLD) are more susceptible to liver ischemia-reperfusion injury (LIRI), complicating liver surgery outcomes. This study aimed to uncover shared hub genes and mechanisms linking LIRI and MASLD to enhance donor liver utilization and improve prognosis. Using liver transplantation and MASLD datasets from the Gene Expression Omnibus, we applied Linear Models for Microarray Data and weighted gene co-expression analysis to identify differentially expressed genes and key module genes. Further analysis involved Gene Ontology, KEGG, and machine learning to pinpoint common hub genes and pathways. We identified 5,920 differentially expressed genes in liver datasets and 8,978 across LIRI and MASLD datasets. 71 shared hub genes were associated with pathways like MAPK signaling. Key genes, ADRB2 and CCL2, exhibited correlated mRNA expression in both datasets and human liver tissues. Hypoxia-reoxygenation in MASLD models elevated CCL2 levels and reduced ADRB2 expression. These genes showed strong diagnostic potential (AUC, 0.97). CCL2 knockdown reduced, while ADRB2 knockdown increased, MASLD cells’ H/R injury sensitivity. Immune infiltration analysis revealed increased immune cell activity, particularly correlations between M0/M2 macrophages and NK cells/mast cells. ADRB2 and CCL2 were identified as crucial biomarkers, potentially explaining MASLD patients’ heightened vulnerability to LIRI during liver transplantation.
Similar content being viewed by others

Introduction
Liver diseases cause nearly two million deaths worldwide, imposing a significant burden on global health and the economy1. Liver transplantation is considered the most effective treatment for end-stage liver disease and provides hope for treatment to patients where other treatments are not effective1. Further, laparoscopic and robotic partial hepatectomies are used to excise both benign and malignant liver tumors2. Despite surgical intervention, liver ischemia-reperfusion injury (LIRI) remains a significant challenge. LIRI manifests as a physiological phenomenon in which blood flow to the liver is momentarily obstructed. However, upon restoration, liver injury is exacerbated. This shows the complexity of surgical management of liver diseases and highlights the need for improving our understanding and identifying innovative strategies to mitigate the impact of LIRI3. During partial hepatectomy, intermittent occlusion and release of the porta hepatis are commonly used to minimize intraoperative bleeding and to enhance surgical safety4. This disconnection and subsequent reconstruction of the portal vein and hepatic artery are essential steps in liver transplantation. Consequently, LIRI is an inevitable complication of the surgical process. Therefore, it is imperative to elucidate the underlying mechanisms and to develop more effective treatments that promote perioperative recovery in patients undergoing liver surgery.
In clinical practice, patients exhibit varying degrees of tolerance to LIRI owing to the differences in their underlying health conditions. Previous studies have demonstrated that the severity of hepatic steatosis influences tolerance to LIRI, and increases the risk of perioperative liver failure after transplantation5. Hence, it is typically recommended that the degree of fat change in the donor liver be less than 30% to mitigate the risk of adverse outcomes6. An increasing prevalence of metabolic dysfunction-associated steatotic liver disease (MASLD) has attracted widespread attention. A meta-analysis showed that approximately 32.4% of the global population has MASLD7. Moreover, this growing incidence of MASLD will increase the demand for liver transplantation. However, the supply of donor livers is limited, which creates a significant gap in the demand and availability of suitable organs. Multiple studies have confirmed that MASLD increases hepatic susceptibility to LIRI, which undoubtedly exacerbates the shortage of available livers for transplantation8;9. Therefore, there is an urgent need to investigate common molecular pathways and key genes underlying MASLD and LIRI. Pathophysiological processes such as inflammation, apoptosis, and oxidative stress are recognized as common determinants of both LIRI and MASLD severity. However, whether these processes represent the true drivers of heightened hepatic susceptibility to IRI in MASLD, and the detailed underlying mechanisms, remain unclear. In this study, we performed integrative analysis of gene sets associated with both IRI and MASLD to screen for key genes and pathways with dual significance in these two conditions, aiming to elucidate the biological mechanisms potentially contributing to MASLD-related IRI hypersensitivity. Understanding these mechanisms could help develop therapies and the lead to optimized use of marginal donor livers, ultimately improving patient outcomes.
-
Currently, many studies have achieved research objectives such as exploring the pathogenesis of diseases or screening molecular markers by screening hub genes through bioinformatics analysis.
-
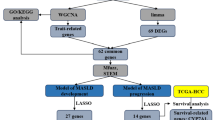
Hub genes are genes that play an important overall regulatory role in the disease gene network, and they are of vital significance for understanding the pathogenesis and treatment of diseases. Bioinformatic analysis is a valuable tool for exploring hub genes and molecular mechanisms in various diseases10. Currently, many studies have achieved research objectives such as exploring the pathogenesis of diseases or screening molecular markers by screening hub genes through bioinformatics analysis11;12. Although existing studies have used bioinformatics analysis methods such as Weighted Gene Co - expression Network Analysis (WGCNA) and machine learning to separately identify hub genes related to LIRI and MASLD, the association between the two is often overlooked13;14. In this study, for the first time, hub genes were further screened based on the common differentially expressed genes of LIRI and MASLD to explore the potential mechanisms underlying the high sensitivity of MASLD to LIRI.
Further, we validated the mRNA levels of the hub genes using RT-qPCR. Finally, we analyzed immune cell infiltration within the liver transplant dataset to elucidate the intricate mechanisms associated with LIRI. Overall, our study leveraged bioinformatic tools to uncover the shared molecular mechanisms of LIRI and MASLD, thus providing a novel theoretical foundation for developing appropriate treatment.
Results
Identification of DEGs in LIRI and MASLD
A comprehensive analysis of DEGs in the liver transplantation datasets GSE151648 for LIRI and GSE89632 for MASLD provided valuable insights into the molecular mechanisms of these conditions. Using the R package LIMMA, we identified 5,920 DEGs in the pre-liver and post-liver transplantation datasets, which met the stringent criterion of adjusted P value < 0.05 (Fig. 1a,b). Similarly, analysis of the MASLD dataset GSE89632 revealed 8,978 DEGs, indicating significant transcriptional changes associated with MASLD pathogenesis (Fig. 1c,d). We visualizing the DEGs through volcano plots and heatmaps using R packages “ggplot2” and “Pheatmap.” These graphical representations provided a comprehensive overview of the alterations in gene expression.

Identification of DEGs in datasets related to LIRI and MASLD. (a) Volcano plot of DEGs in the GSE151648 dataset (LIRI). (b) Heatmap of top 30 DEGs in the GSE151648 dataset (LIRI). (c) Volcano plot of DEGs in the GSE89632 dataset (MASLD). (d) Heatmap of top 30 DEGs in the GSE89632 dataset (MASLD).
Weighted gene co-expression network analysis and key module identification
Weighted gene co-expression network analysis (WGCNA) is a powerful method for identifying hub genes from a large pool of genes by clustering them into distinct modules based on similarity of expression patterns15. By partitioning genes into modules, WGCNA facilitates the prioritization of hub genes that are highly connected within the respective modules and are often key regulators or drivers of biological processes. Thus, WGCNA serves as an effective strategy for unraveling complex gene networks and elucidating the molecular mechanisms underlying various biological phenomena, including disease pathogenesis and therapeutic responses. In our study, WGCNA was used to identify the modules that were most correlated with LIRI and MASLD. After determining β = 4 (scale-free R2 = 0.88) for GSE151648 (Fig. 2a,b) and β = 6 (scale-free R2 = 0.73) for GSE89632 (Fig. 3a,b), 19 modules in GSE151648 (Fig. 2d,e) and 16 modules in GSE89632 (Fig. 3d,e) were identified, each designated with a distinct color. The dendrograms of clustering for the two datasets are shown in Figs. 2c and 3c. Through correlation analysis between the modules and phenotypes, the blue module genes in GSE151648 exhibited the highest correlation with LIRI (r = 0.88, p = 1.1e − 26) (Fig. 2f,g), whereas the brown module genes in GSE89632 showed the highest correlation with MASLD (r = 0.82, p = 3.5e − 16) (Fig. 3f,g). Using the cutoff criterion |MM| >0.8, 283 hub genes in the blue module and 236 hub genes in the brown module were identified for further analysis. These hub genes may play pivotal roles in the development of LIRI and MASLD and provide valuable insights into disease mechanisms and potential therapeutic targets.

Identification of key module genes by WGCNA in GSE151648. (a,b) β = 4 is determined as the soft threshold with the combined analysis of scale independence and average connectivity. (c) Clustering dendrogram of the samples in GSE151648. (d) Gene co-expression modules represented by different colors under the gene tree. (e) Eigengene adjacency heatmap. (f) Heatmap of the correlation between module genes and LIRI. (g) Correlation of module membership and gene significance in the blue module.

Identification of key module genes by WGCNA in GSE89632. (a,b) β = 6 is determined as the soft threshold with the combined analysis of scale independence and average connectivity. (c) Clustering dendrogram of the samples in GSE89632. (d) Gene co-expression modules represented by different colors under the gene tree. (e) Eigengene adjacency heatmap. (f) Heatmap of the correlation between module genes and MASLD. (g) Correlation of module membership and gene significance in the brown module.
Biological processes and pathway enrichment analysis of common hub genes
After meticulously screening the hub genes within the modules associated with LIRI and MASLD, we identified 71 common key genes in the intersection set (Fig. 4a). To unravel the intricate mechanisms underlying the influence of these genes on disease progression, we conducted comprehensive Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses to elucidate their biological functions, cellular components, molecular functions, and involvement in signaling pathways. The results were visually represented using bubble maps highlighting the top 10 enriched terms. Bubble maps revealed significant enrichment in terms of “negative regulation of cellular processes” and “positive regulation of metabolic processes” within the biological function category (Fig. 4b). In the cellular components, the genes exhibited enrichment in crucial areas such as “cell surface” and the “collagen-containing extracellular matrix,” indicating the pivotal roles in the cellular architecture and extracellular matrix dynamics (Fig. 4c). Within the molecular functions category, notable enrichment was observed in “molecular function regulator” and “signaling receptor binding,” indicating the involvement of these genes in modulating molecular processes and interactions with signaling receptors (Fig. 4d). Furthermore, enrichment analysis of the KEGG pathways revealed various signaling pathways, and the MAPK signaling pathway emerged as the most significantly enriched pathway (Fig. 4e). These findings highlight the intricate interplay of genes associated with LIRI and MASLD and the key signaling pathways such as MAPK, in disease progression. These results are consistent with that of previous studies on MASLD and LIRI reinforcing the robustness and reliability of our analysis16;17.

Enrichment analysis of common key genes in LIRI and MASLD. (a) Venn diagram illustrating the selection of common key genes between LIRI and MASLD. (b–d) GO analysis of common key genes. (e) KEGG analysis of common key genes.
Utilizing multiple machine learning approaches for screening of hub genes and evaluation of diagnostic value
We utilized LASSO regression and Support Vector Machine (SVM) to conduct an additional screening of the 71 common key genes. In the LASSO regression machine learning method, we set the λ value to 0.05, and identified nine potential genes, namely MAP1LC3B, ADRB2, KLF6, BTG3, CCL2, TNFRSF12A, PPP1R15A, MAFF, and HAS2 (Fig. 5a,b). Using the SVM machine learning approach, we screened eight potential key genes, namely ETS2, CCL2, EMP1, TGFB3, KIAA0040, ADM, DUSP5, and ADRB2 (Fig. 5c). ADRB2 and CCL2 were identified as the intersecting hub genes using both methods (Fig. 5d). Subsequently, a receiver operating characteristic (ROC) analysis of the aforementioned genes was done to derive the area under the curve (AUC) values and the corresponding 95% confidence intervals (CI). Both ADRB2 (AUC 0.97, 95% CI 1.00–0.94) and CCL2 (AUC 0.97, 95% CI 1.00–0.94) exhibited significant diagnostic potential for LIRI demonstrating robust discriminative power in distinguishing between affected and unaffected states (Fig. 5e,f).

Machine learning of common key genes in LIRI and MASLD. (a,b) Nine genes were identified by Lasso regression. (c) Eight genes were identified by SVM machine learning. (d) Venn diagram illustrating the common hub genes obtained with two machine learning. (e,f) ROC analysis of ADRB2 and CCL2.
Expression changes and functional verification of CCL2 and ADRB2 in LIRI and MASLD
We collected liver tissue samples from patients who underwent partial hepatectomy for hemangiomas and performed RT-qPCR. The results revealed a significant increase in the CCL2 and ADRB2 levels after LIRI (Fig. 6a, b). In comparison, the liver tissues of patients with MASLD exhibited significantly decreased mRNA levels of CCL2 and ADRB2 compared to the control group (Fig. 6c, d). These findings were consistent with the findings from the Gene Expression Omnibus (GEO) database (Fig. 6e-h).

Expression changes and functional exploration of CCL2 and ADRB2 in LIRI and MASLD. (a,b) mRNA expression changes of CCL2 and ADRB2 in human liver tissues undergoing LIRI (n = 6/group). (c,d) Comparison of CCL2 and ADRB2 mRNA levels between MASLD and control groups in human liver tissues (n = 4/group). (e,f) Expression changes of CCL2 and ADRB2 in the GSE151648 dataset (n = 40/group). (g,h) Expression changes of CCL2 and ADRB2 in the GSE89632 dataset (Con, n = 24; MASLD, n = 39). (i) Comparison of cell viability between the control group and MASLD group (n = 4/group). (j,k) Changes in mRNA expression of CCL2 and ADRB2 in vitro (n = 3/group). (l,m) Efficiency verification of RT-qPCR for siRNAs targeting CCL2 and ADRB2 (n = 3/group). (n) Comparison of cell viability between siNC and siCCL2-2 groups under H/R in normal and MASLD cell models (n = 5/group). (o) Comparison of cell viability between siNC and siADRB2-2 groups under H/R in normal and MASLD cell models (n = 5/group).
Subsequently, we established a cellular model of MASLD by treating hepatocytes with oleic acid and palmitic acid, followed by H/R treatment. The results of the CCK8 assay indicated that the sensitivity of steatotic hepatocytes to H/R injury was significantly higher than that of the control group (Fig. 6i). RT-qPCR analysis revealed that CCL2 levels in the MASLD group were significantly higher than those in the control group under the same H/R treatment conditions. Furthermore, the H/R-induced elevation of ADRB2 levels was mitigated in the MASLD group (Fig. 6j,k; Supplementary Fig. S1a,b). To investigate the roles of CCL2 and ADRB2 in this process, we constructed siRNAs targeting CCL2 and ADRB2. The knockdown efficiency was verified by RT - qPCR, and the siRNAs with the most significant knockdown efficiency were selected for subsequent experiments (Fig. 6l,m). Subsequently, the results of the CCK8 assay showed that knocking down CCL2 could significantly reduce the sensitivity of the MASLD cell model to H/R injury, while knocking down ADRB2 had the opposite effect (Fig. 6n,o). This indicates that within the context of MASLD, there may be alterations in the expression of ADRB2 and CCL2 during the LIRI process, which could subsequently affect the severity of LIRI.
To further verify the reliability of our research findings, we selected additional LIRI dataset (GSE15480) and MASLD dataset (GSE37031) for further validation. The results of the differential analysis showed that the expression changes of CCL2 and ADRB2 in the validation set were consistent with our above-mentioned research results (Supplementary Fig. S2a-d). Moreover, the ROC curves indicated that CCL2 and ADRB2 have good diagnostic effects on LIRI (Supplementary Fig. S2e, f), which further enhances the credibility of this study.
Immune cell infiltration analysis
CCL2 plays a crucial role in immune responses, inflammation, and tissue repair18. Additionally, CCL2 also regulates the tumor microenvironment and promotes tumor growth, metastasis, and angiogenesis19,20,21. ADRB2 is a G-protein-coupled receptor involved in immune regulation22. A previous study showed that ADRB2 gene polymorphisms influence individual patterns of immune response, further affecting the process of norepinephrine-mediated immunosuppression23. Therefore, we analyzed the LIRI dataset from the perspective of immune cell infiltration to elucidate the underlying mechanisms.
The distribution of immune cell infiltration across the samples is displayed in a stacked graph (Fig. 7a). As shown in the box plot, we observed a significant increase in the proportion of infiltrating memory B cells, CD4+-activated memory T cells, resting NK cells, monocytes, M0 macrophages, activated dendritic cells, activated mast cells, and eosinophils after LIRI. Conversely, the infiltration of native B cells, CD4+ T cells, follicular helper T cells, activated NK cells, M1 and M2 macrophages, resting dendritic cells, and resting mast cells was decreased (Fig. 7b). Subsequently, correlation analysis of the 22 immune cell types was performed. The results showed a negative correlation between M0 macrophages and M2 macrophages (r = − 0.55), and resting NK cells were positively correlated with activated mast cells (r = 0.54) (Fig. 7c). From the results of immune infiltration analysis, it is evident that LIRI led to activation and infiltration changes in various immune cells. These findings suggest that immunotherapy might be a new strategy for alleviating LIRI.

Immune cell infiltration analysis in LIRI. (a) The proportion of different immune cells in different samples. (b) Comparison of the proportion of different immune cells between pre-liver and post-liver transplantation visualized by box plot. (c) Correlation of different immune cell type compositions. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, no significance.
Discussion
MASLD is an important contributor to the development of end-stage liver disease. Further, LIRI exacerbates liver damage, contributing to liver transplantation failure. Alleviating LIRI can increase the utilization rate of marginal donor livers. Additionally, patients with MASLD demonstrate increased susceptibility to LIRI-induced injury. Therefore, exploring the pathogenic mechanisms underlying MASLD and LIRI is of paramount importance. Currently, multiple treatment methods are used to alleviate LIRI including pharmacological interventions such as anti-inflammatory agents and antioxidants, and preconditioning techniques such as ischemic preconditioning24,25. Surgical interventions such as ischemic postconditioning and remote ischemic conditioning have also been explored26. However, the effectiveness of all current treatment methods is unsatisfactory.
Immunotherapy has become a research hotspot for the treatment of various diseases, especially malignant tumors. For example, PD-1 and PD-L1 immune checkpoint therapies have demonstrated significant clinical efficacy in various cancers such as liver, lung, and kidney cancers. PD-1 and PD-L1 inhibitors can relieve immune suppression of tumor cells by T cells, thereby restoring T cell activity and promoting the immune system to attack tumor cells27. Therefore, PD-1 and PD-L1 immune checkpoint therapies have shown great potential for the treatment of various cancers and have provided new treatment options. Several studies have investigated immune mechanisms involved in LIRI. Macrophages are integral components of the immune response during LIRI progression28. Under ischemic and hypoxic conditions, hepatocyte membranes are disrupted, leading to the release of damage-associated molecular patterns that subsequently trigger polarization of macrophages towards the M1 phenotype. In the later stages of reperfusion, macrophages polarize towards the M2 phenotype and secrete anti-inflammatory cytokines, thereby alleviating inflammation29. Several studies have elucidated the mechanisms underlying LIRI based on macrophage polarization. Additionally, activation and recruitment of CD4 + T cells can influence the severity of LIRI30. Therefore, exploring new and effective treatments for alleviating LIRI from an immunotherapeutic perspective is highly meaningful.
In our previous work, we used bioinformatics analysis to screen for genes associated with m6A/m7G/m5C/m1A during the LIRI process, and identified the pathways controlled by these genes31. To investigate why patients with MASLD were more sensitive to LIRI and explore the potential associations between LIRI and other liver diseases, this study focused on the commonalities between the disease processes of MASLD and LIRI. We performed bioinformatic analysis on two datasets related to LIRI and MASLD, GSE151648 and GSE89632. These analyses identified the pathways and genes common to both these diseases. Traditional gene screening methods, such as simple differential expression analysis, usually can only identify differentially expressed genes based on pre-set statistical thresholds. This approach makes it difficult to consider the complex interaction relationships among genes. Machine learning can mine the complex nonlinear relationships of genes related to LIRI and MASLD. Methods like LASSO regression and SVM can automatically capture such relationships, which is beyond the reach of traditional single methods. Moreover, these machine learning methods have been widely applied in the screening process of hub genes for various diseases. Therefore, after obtaining the common key gene groups of LIRI and MASLD through WGCNA, we further conduct dimensionality reduction screening by means of machine learning. Hub genes ADRB2 and CCL2, which were previously confirmed to be closely associated with immune responses, were identified as shared hub genes that affected both LIRI and MASLD. The ROC analysis showed that these two genes had good diagnostic ability for LIRI. Therefore, detecting the expression levels of ADRB2 and CCL2 in donor liver tissues might help predict the severity of LIRI injury and provide timely treatment measures to avoid adverse outcomes.
Currently, some studies have shown that ADRB2 can influence the proliferation and apoptosis of T cells as well as the inflammatory phenotype of macrophage32,33. As a chemokine, CCL2 can affect the infiltration of monocytes/macrophages and myeloid suppressor cells, as well as the differentiation of B cells34,35,36. Based on our immune infiltration analysis, it is evident that various immune cells undergo significant changes during LIRI. Therefore, we speculate that CCL2 and ADRB2 may also affect the severity of LIRI in vivo by influencing the infiltration of immune cells. This speculation needs to be further explored in future animal experiments, which may provide new ideas for the research and development of immunotherapy for LIRI.
There are limitations to this study. Although we have confirmed the expression changes of CCL2 and ADRB2 through databases and cytological experiments, and conducted functional verification in in-vitro experiments, there is still a lack of in-vivo experiments to further explore the effects of CCL2 and ADRB2 on non - parenchymal cells such as immune cells during this process. In future research, we will continue to investigate the impact of these hub genes on the severity of LIRI in MASLD. We believe continuing research in this area will help in the development of novel therapeutic approaches to enhance the utilization rate of donor livers and mitigate the severity of LIRI in patients with MASLD.
Conclusions
Our results demonstrated that LIRI and MASLD are associated with multiple common genes. Using machine learning, we identified ADRB2 and CCL2 as hub genes that exhibited good diagnostic capabilities for LIRI. These genes affect the tolerance of hepatocytes to H/R injury and may be involved in regulating the infiltration of immune cells during the LIRI process.
Materials and methods
Data acquisition and processing
Liver transplantation (GSE151648)37 and MASLD (GSE89632)38 datasets were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The GSE151648 dataset included 40 patients who underwent liver transplantation. For high-throughput sequencing, liver tissues were collected from each patient before and after transplantation. The GSE89632 dataset contained expression profiles of liver tissues obtained from 39 patients with MASLD and 24 healthy individuals. The inclusion and exclusion criteria for patients were similar with the protocols of the original studies37,38. After obtaining the data, we transform non-standardized data with Log2(x + 1) to compress the dynamic range and stabilize variance. Then, we use the normalizeBetweenArrays function in the Limma R package for standardization. This makes gene expression data of different samples comparable. After completing the data standardization, to ensure the consistency and accuracy of gene information and facilitate subsequent in-depth analysis, we utilized the information from relevant platforms to uniformly convert various types of gene IDs into the universal Gene symbol. During the processing, in case of duplicate gene names, to avoid data deviation, we calculated the average value of the relevant data. In addition, after conducting the differential analysis, we removed the information related to genes containing missing values, further optimizing the data quality and ensuring the reliability of the analysis results.
Identification of differentially expressed genes
We used the “LIMMA” package in R to identify DEGs. In the analysis, we controlled the false discovery rate (FDR) with the Benjamini - Hochberg method at a 0.05 significance level, and considered genes with an adjusted p - value < 0.05 and |log2 fold change| > 1 as DEGs. Subsequently, we leveraged the “ggplot2” package to craft a volcano plot, vividly visualizing the significance and fold - change of differentially expressed genes. Meanwhile, the “Pheatmap” package was employed to generate a heatmap, which presented the gene expression patterns across samples in an intuitive manner.
Weighted gene co-expression network analysis
We calculated the median absolute deviation (MAD) for gene expression variability by filtering out the lowest 50% of genes. By employing WGCNA’s “goodSamplesGenes” function in R package, we refined our dataset by removing outlier genes and samples. With β as the soft-thresholding power, we prioritized significant gene correlations. The adjacency matrix was transitioned into a topological overlap matrix (TOM) to capture gene connectivity and the corresponding dissimilarity (1-TOM) value was derived. We consolidated the genes into coherent modules, each with at least 30 members, using hierarchical clustering with TOM-based metrics. High-connectivity genes (|MM| >0.8) were identified as potential hub genes within influential modules.
Enrichment analysis
We utilized the “clusterProfiler” R package to perform GO and KEGG analyses with the aim of exploring gene functions and associated pathways.
Machine learning
We applied LASSO regression using “glmnet” in R package and optimized the model through five-fold cross-validation. Additionally, the R package e1071 was used to conduct SVM learning, which enabled the extraction of the top genes from the analysis. By employing these complementary methods, we aimed to identify and prioritize key genes associated with the studied conditions.
Analysis of immune infiltration
We used CIBERSORT to analyze immune cell infiltration in the liver transplantation dataset to gain insights into the immune cell composition of liver transplantation samples. This provided valuable information for understanding immune response dynamics in the context of LIRI.
Clinical sample collection
We collected liver tissue samples from 10 patients who underwent partial hepatectomy for hepatic hemangioma at the First Affiliated Hospital of Harbin Medical University between December 2023 and March 2024. Patients with poor baseline conditions and significant preoperative abnormalities in liver and kidney functions were excluded from the study.
Cell culture and treatment
AML12 cells (Cell Bank of the Chinese Academy of Sciences, Shanghai, China) were cultured in AML12 cells complete medium (Gibco, USA) at 37 °C constant temperature and 5% CO2/water-saturated incubator. The WRL68 human normal cell line was obtained from the Chinese Academy of Sciences’ Type Culture Collection (Shanghai). It was cultured in DMEM with 10% fetal bovine serum in a 37 °C, 5% CO₂, water - saturated incubator. The cells were treated with a mixture of 0.5 mmol/L oleic acid (OA) and palmitic acid (PA) (oleic: palmitic = 2:1) for 24 h to establish an MASLD cellular model39. After incubation for 6 h in a CO2 incubator with 1% O2, 5% CO2, and 94% N2, the cells were transferred to a normal culture incubator for 6 h to establish the cellular hypoxia–reoxygenation (H/R) model40.
SiRNA construction and transfection
Gene silencing was achieved using siRNAs custom - designed and synthesized by HaiXing Biotechnology (Suzhou, China). The specific sequences utilized can be found in Supplementary Table S2. SiRNA transfection into hepatocytes was carried out in strict accordance with the instructions provided for Lipofectamine 3000 (Invitrogen, USA).
Cell viability determination
Hepatocyte viability was assessed using the CCK-8 assay (Dojindo molecular technologies, CK04-13) according to the manufacturer’s instructions.
RT-qPCR
An RNA extraction kit (AxyPrep multisource total rNA miniprep kit, Axygen®) was used for RNA extraction from liver tissue samples and cells. Subsequently, the extracted RNA was utilized as a template for cDNA synthesis with a reverse transcription kit (ReverTra Ace™ qPCR RT master mix, TOYOBO.inc, Japan). The primer sequences were custom-designed and manufactured by General Biology (Chuzhou, China; Supplementary Table S1). RT-qPCR analysis was done using a pre-made mix (FastStart™ Universal SYBR® Green Master, Rox), and data analysis conducted using the 2−ΔΔCt method, with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the reference gene.
Statistical analysis
We used R software version 4.3.1 for bioinformatics and statistical analyses. Differences between groups were assessed using the Student’s t-test. Pearson’s correlation test was used to assess the associations between variables. P values < 0.05 were considered statistically significant.
Data availability
Liver transplantation (GSE151648, GSE15480) and MASLD (GSE89632, GSE37031) datasets were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/).
References
Bhat, M., Rabindranath, M., Chara, B. S. & Simonetto, D. A. Artificial intelligence, machine learning, and deep learning in liver transplantation. J. Hepatol. 78, 1216–1233 (2023).
Kokudo, N., Takemura, N., Ito, K. & Mihara, F. The history of liver surgery: achievements over the past 50 years. Ann. Gastroenterol. Surg. 4, 109–117 (2020).
Papadopoulos, D., Siempis, T., Theodorakou, E. & Tsoulfas, G. Hepatic ischemia and reperfusion injury and trauma: current concepts. Arch. Trauma. Res. 2, 63–70 (2013).
Mownah, O. A. & Aroori, S. The pringle maneuver in the modern era: A review of techniques for hepatic inflow occlusion in minimally invasive liver resection. Ann. Hepatobiliary Pancreat. Surg. 27, 131–140 (2023).
Majumdar, A. & Tsochatzis, E. A. Changing trends of liver transplantation and mortality from Non-Alcoholic fatty liver disease. Metabolism 111S, 154291 (2020).
Jackson, K. R. et al. Temporal trends in utilization and outcomes of steatotic donor livers in the united States. Am. J. Transpl. 20, 855–863 (2020).
Riazi, K. et al. The prevalence and incidence of Nafld worldwide: A systematic review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 7, 851–861 (2022).
Yu, S. et al. Nonalcoholic steatohepatitis critically rewires the Ischemia/Reperfusion-Induced dysregulation of cardiolipins and sphingolipids in mice. Hepatobil Surg. Nutr. 12, 3–19 (2023).
Liu, R. et al. Zbp1-Mediated apoptosis and inflammation exacerbate steatotic liver ischemia/reperfusion injury. J. Clin. Invest. 134 (2024).
Gauthier, J., Vincent, A. T., Charette, S. J. & Derome, N. A brief history of bioinformatics. Brief. Bioinform. 20, 1981–1996 (2019).
Chen, H. et al. Analysis of necroptosis-related prognostic genes and immune infiltration in idiopathic pulmonary fibrosis. Front. Immunol. 14, 1119139 (2023).
Chen, Y. et al. Identification of core Immune-Related genes Ctsk, C3, and Ifitm1 for diagnosing Helicobacter Pylori Infection-Associated gastric Cancer through transcriptomic analysis. Int. J. Biol. Macromol. 287, 138645 (2025).
Chen, Y., Zhang, J. & Li, F. Inhibitory role of remifentanil in hepatic Ischemia-Reperfusion injury through activation of Fmol/Parkin signaling pathway: A study based on network Pharmacology analysis and High-Throughput sequencing. Phytomedicine 128, 155300 (2024).
He, R., Guan, C., Zhao, X., Yu, L. & Cui, Y. Expression of immune related genes and possible regulatory mechanisms in different stages of Non-Alcoholic fatty liver disease. Front. Immunol. 15, 1364442 (2024).
Langfelder, P. & Horvath, S. Wgcna: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Fang, Z. et al. Short-Term Tamoxifen administration improves hepatic steatosis and glucose intolerance through Jnk/Mapk in mice. Signal. Transduct. Target. Ther. 8, 94 (2023).
Yu, B. et al. Mapk signaling pathways in hepatic ischemia/reperfusion injury. J. Inflamm. Res. 16, 1405–1418 (2023).
Singh, S., Anshita, D. & Ravichandiran, V. Mcp-1: function, regulation, and involvement in disease. Int. Immunopharmacol. 101, 107598 (2021).
Jin, J. et al. Ccl2: an important mediator between tumor cells and host cells in tumor microenvironment. Front. Oncol. 11, 722916 (2021).
Xie, M. et al. Fgf19/Fgfr4-Mediated elevation of Etv4 facilitates hepatocellular carcinoma metastasis by upregulating Pd-L1 and Ccl2. J. Hepatol. 79, 109–125 (2023).
Zeng, H. et al. Cancer-Associated fibroblasts facilitate premetastatic niche formation through Lncrna Snhg5-Mediated angiogenesis and vascular permeability in breast Cancer. Theranostics 12, 7351–7370 (2022).
Wang, Y. & Jiang, S. The role of Adrb2 gene polymorphisms in malignancies. Mol. Biol. Rep. 48, 2741–2749 (2021).
Stolk, R. F. et al. The impact of Adrb2 polymorphisms on immune responses and Norepinephrine-Induced immunosuppression. J. Leukoc. Biol. 113, 84–92 (2023).
Yan, Y. et al. Ischemic preconditioning increases Gsk-3Β/Β-Catenin levels and ameliorates liver ischemia/reperfusion injury in rats. Int. J. Mol. Med. 35, 1625–1632 (2015).
Li, J. et al. Isolongifolene alleviates liver ischemia/reperfusion injury by regulating Ampk-Pgc1Α signaling Pathway-Mediated inflammation, apoptosis, and oxidative stress. Int. Immunopharmacol. 113, 109185 (2022).
Costa, F. L. et al. Combined remote ischemic perconditioning and local postconditioning on liver Ischemia-Reperfusion injury. J. Surg. Res. 192, 98–102 (2014).
Yi, M. et al. Combination strategies with Pd-1/Pd-L1 Blockade: current advances and future directions. Mol. Cancer. 21, 28 (2022).
Wang, L. et al. Adar1 regulates macrophage polarization and is protective against liver ischemia and reperfusion injury. Immunobiology 229, 152777 (2024).
Hirao, H., Nakamura, K. & Kupiec-Weglinski, J. W. Liver Ischaemia-Reperfusion injury: A new Understanding of the role of innate immunity. Nat. Rev. Gastroenterol. Hepatol. 19, 239–256 (2022).
Al, M. S. et al. Inhibition of Γδ-Tcr or Il17a reduces T-Cell and neutrophil infiltration after ischemia/reperfusion injury in mouse liver. J. Clin. Med. 12 (2023).
Meng, Z. et al. A comprehensive analysis of M6a/M7G/M5C/M1a-Related gene expression and immune infiltration in liver Ischemia-Reperfusion injury by integrating bioinformatics and machine learning algorithms. Eur. J. Med. Res. 29, 326 (2024).
Ajmal, I. et al. Intrinsic Adrb2 Inhibition improves Car-T cell therapy efficacy against prostate Cancer. Mol. Ther. 32, 3539–3557 (2024).
Hasegawa, S. et al. Activation of sympathetic signaling in macrophages blocks systemic inflammation and protects against renal Ischemia-Reperfusion injury. J. Am. Soc. Nephrol. 32, 1599–1615 (2021).
Li, X. et al. Targeting of Tumour-Infiltrating macrophages via Ccl2/Ccr2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 66, 157–167 (2017).
Huang, B. et al. Ccl2/Ccr2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 252, 86–92 (2007).
Yang, L. et al. Ccl2 regulation of Mst1-Mtor-Stat1 signaling Axis controls Bcr signaling and B-Cell differentiation. Cell. Death Differ. 28, 2616–2633 (2021).
Sosa, R. A. et al. Disulfide High-Mobility group box 1 drives Ischemia-Reperfusion injury in human liver transplantation. Hepatology 73, 1158–1175 (2021).
Arendt, B. M. et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic N-3 and N-6 polyunsaturated fatty acids. Hepatology 61, 1565–1578 (2015).
Gao, R. et al. Mitochondrial pyruvate carrier 1 regulates fatty acid synthase lactylation and mediates treatment of nonalcoholic fatty liver disease. Hepatology 78, 1800–1815 (2023).
Wang, C. et al. Lncrna Hnf4Αos exacerbates liver ischemia/reperfusion injury in mice via Hnf4Αos/Hnf4Α Duplex-Mediated Pgc1Α suppression. Redox Biol. 57, 102498 (2022).
Acknowledgements
Not applicable.
Funding
This work was supported by the Research Fund of the National Natural Science Foundation of China (82370643), Natural Science Foundation of Heilongjiang Province of China (LC2018037), Chen Xiaoping Foundation for the Development of Science and Technology of Hubei Province (CXPJJH11900001-2019349).
Author information
Authors and Affiliations
Contributions
Yong Ma: Conceptualization; Yongzhi Zhou: Formal analysis, visualization, writing-original draft; Bing Yin, Yang Yang: Methodology; Zhongyu Li and Zhanzhi Meng: Data curation; Shounan Lu, Baolin Qian, Xinglong Li, Yongliang Hua , Hongjun Yu and Yao Fu: Writing- Reviewing and Editing. All authors have confirmed the final.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All liver tissue samples were obtained with the informed consent of the patients. This study was conducted in accordance with the guidelines mentioned on Declaration of Helsinki and was approved by the Ethics Committee of the First Affiliated Hospital of Harbin Medical University.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, Y., Yin, B., Yang, Y. et al. Identifying shared hub genes in LIRI and MASLD through bioinformatics analysis and machine learning. Sci Rep 15, 18532 (2025). https://doi.org/10.1038/s41598-025-01609-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01609-8
