
Abstract
Ectomycorrhizae (ECM) play a critical role in enhancing plant growth and health. However, the influence of artificially established ectomycorrhizal symbioses on the structure and function of rhizosphere microbial communities remains inadequately understood. In this study, a symbiotic relationship between Hebeloma hiemale and Quercus mongolica was established to investigate the influence of ECM on soil microbial communities in the rhizosphere of the host plant. High-throughput sequencing revealed that H. hiemale inoculation altered the evenness of both the fungal and bacterial communities and reduced the diversity of the bacterial community relative to the blank control. In particular, several bacterial genera with an enhanced capacity for nutrient cycling, contaminant degradation, and host plant protection were enriched following H. hiemale inoculation. Shifts in fungal community structure suggest potential benefits for the host plant, including reduced cadmium uptake, enhanced mercury remediation, and increased protection against pathogens. Our results highlight the complex interactions between ECM and rhizosphere microbial communities to enable a better understanding of the importance of multi-species relationships in plant–microbe symbioses and their ecological implications.
Similar content being viewed by others


Introduction
Mycorrhizae are well-known mutualistic symbioses between soil fungi and host plant roots. They are commonly divided into two types: ectomycorrhizae (ECM) and endomycorrhizae1,2,3. ECM symbioses are significant components of forest ecosystems and play essential ecological roles in enhancing plant growth and health4,5. Hebeloma hiemale is an ECM-forming fungus that promotes plant growth6,7. ECM acquire carbon from the host plant and enhances the uptake of essential nutrients (e.g., nitrogen and phosphorus) and water by the host plant8,9. ECM symbioses are artificially established by the inoculation of host-beneficial ECM, offering a promising strategy for enhancing plant growth and resilience in degraded environments10,11. This approach is a sustainable alternative to conventional agricultural and forestry practices12. For instance, Quercus mongolica, an ecologically and economically valuable species13,14, can successfully establish an artificial ECM symbiosis when inoculated with Gomphidius viscidus15. However, the influence of artificially established ECM symbioses on microbial communities in the rhizosphere of Q. mongolica remains inadequately understood.
The microbial community is an essential component of plant life16,17,18. Based on their influence on plants, microbial communities are classified as beneficial, neutral, or harmful19,20. Beneficial microbes can enhance plant growth and adaptation21,22. On the other hand, harmful microbes, including plant pathogens, can adversely affect plant growth23,24,25. ECM not only form mutualistic symbioses with the host plant but also influences the rhizosphere microbial community26,27. For instance, Tricholoma bakamatsutake significantly alters the composition of the soil microbial community by suppressing the abundance of other fungal genera, such as Russula and Penicillium, while increasing the abundance of plant growth-promoting bacteria, such as Solirubrobacter and Streptomyces28.
To investigate the influence of ECM on microbial communities in the rhizosphere of the host plant, Q. mongolica was artificially inoculated with H. hiemale. High-throughput sequencing was conducted to assess bacterial (16 s rDNA) and fungal (ITS rDNA) sequences from the rhizosphere microbial community. This study aimed to explore the influence of ECM on fungal and bacterial communities in the rhizosphere of Q. mongolica.
Materials and methods
Description of ectomycorrhizal synthesis and sampling strategy
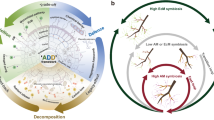
The experiment was conducted in a greenhouse at Shenyang Agricultural University in Shenyang, Liaoning Province, China (41°80’N 123°45’E). The method followed in this study was adopted from previous studies with some modifications29,30,31,32. Q. mongolica seeds (obtained from the Laotudingzi National Nature Reserve, Benxi, China) were surface-sterilized with 30% H2O2 for 4 h and washed thrice with distilled water. The soil was autoclaved at 121 °C for 90 min. The treated seeds were sown in a plastic container filled with autoclaved soil. After 30 days, the seedlings were transplanted into pots containing 1 L of sterilized soil. Soil pH was adjusted to 6.5 6. The experimental procedure is illustrated in Fig. 1. There were three experimental groups, each containing 20 seeding repeats. In the Hh1 and Hh2 groups, each seedling received 5 mL of H. hiemale at a concentration of 107 and 108 spores/mL, respectively. None of the seedlings in the CK group received H. hiemale spores. The three groups were maintained in a greenhouse under the same environmental conditions, watered every 3 days, and no fertilizer was added to the pots. After 150 days, the root tips of Q. mongolica were harvested and soil samples were obtained. Ectomycorrhizal synthesis was assessed using a Nikon SMZ25 stereomicroscope and via ITS rDNA sequence analysis (Fig. 2; GenBank accession number: PP897331). In the Hh1 and Hh2 groups, 14 and 18 seedlings, respectively, showed successfully ectomycorrhizal colonization. In the CK group, all seedlings remained uncolonized by ECM. The rhizospheres were carefully collected from each pot by gently brushing the root surfaces with a sterile brush after removing the loosely adhering soil by gentle shaking31. For each treatment group, we employed a partial pooling strategy to balance analytical depth with experimental feasibility33,34,35. Within each group, samples from individual seedlings were randomly divided into three subgroups. Specifically, in the Hh1 group (n = 14 successful colonizations), samples were pooled into three analytical replicates with four to five individuals per pool. In the Hh2 group (n = 18 successful colonizations), samples were pooled into three analytical replicates, with six individuals per pool. In the CK group (n = 20), the samples were similarly pooled into three analytical replicates with to six to seven individuals per pool. This pooling approach allowed us to focus on the treatment-level effects while maintaining sufficient biological representation for each group. Pooled samples were subjected to DNA extraction for high-throughput sequencing.

Schematic representation of the experimental procedure.

Root tips of Q. mongolica without H. hiemale (a) and with H. hiemale (b, c, d). Scale bars represent 1000 μm.
DNA extraction, PCR amplification, and illumina miseq sequencing
Total metagenomic DNA from the microbial community was extracted using the E.Z.N.A.® soil DNA kit (Omega Bio-tek, Norcross, GA, U.S.) as per manufacturer’s instructions. The quality of the extracted metagenomic DNA was determined using 1% agarose gel electrophoresis, and the concentration and purity of the DNA was determined using a NanoDrop 2000 (Thermo Scientific, USA).
For bacteria, 16S-specific primers 338 F (5’-ACTCCTACGGGAGGCAGCAG-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) were used to amplify the 16S rRNA gene. For fungi, the primers ITS1 F (5’ -CTTGGTCATTTAGAGGAAGTAA-3’) and ITS2R (5’ -GCTGCGTTCTTCATCGATGC-3’) were used to amplify the ITS gene. The PCR reaction mixture comprised 4 µL of 5 × Fast Pfu buffer, 2 µL of 2.5 mM dNTPs, 0.8 µL of each primer (5 µM), 0.4 µL of Fast Pfu polymerase, 10 ng of template DNA, and ddH2O to a final volume of 20 µL. PCR amplification cycling conditions were as follows: initial denaturation at 95 ℃ for 3 min; followed by 27 cycles of denaturing at 95 ℃ for 30 s, annealing at 55 ℃ for 30 s, and extension at 72 ℃ for 45 s; single extension at 72 ℃ for 10 min; and end of the reaction at 4℃. All samples were amplified in triplicate. The PCR product was extracted from 2% agarose gel and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to manufacturer’s instructions and quantified using a Quantus™ Fluorometer (Promega, USA).
Purified amplicons were pooled in equimolar amounts and paired-end sequencing was performed on an Illumina MiSeq PE300 platform (Illumina, San Diego, CA, USA) according to the standard protocols of Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). The raw sequencing reads were deposited in the NCBI Sequence Read Archive database (BioProject: PRJNA1116842).
Data processing
Quality control of raw paired-end sequences was performed using fastp (version 0.19.6)36. The filtered sequences were then merged using FLASH software (version 1.2.11)37. Operational taxonomic units (OTUs) were clustered using UPARSE (version 7.1)38,39 at 97% similarity and chimeras were removed. To minimize the impact of sequencing depth on subsequent alpha and beta diversity analyses, the sequence number was rarefied to the minimum number of sequences across all samples. Taxonomic annotation of OTUs was performed using the RDP classifier (version 2.11)40 against the Silva 16 S rRNA (Release 138)41 and Unite ITS databases (Release 8.0)42 with a confidence threshold of 70%. The community composition of each sample was summarized at different taxonomic levels. Functional predictions were performed using PICRUSt2 (version 2.2.0)43.
Statistical analysis
Data analysis was performed using Meguiar’s BioCloud platform (https://cloud.majorbio.com) and R software (version 4.3.3). Alpha diversity indices, including Chao and Shannon’s indices, were calculated using the Mothur software44. Intergroup variations in alpha diversity indices were assessed using the Wilcoxon rank-sum test. Non-metric multidimensional scaling (NMDS) analysis, based on the Bray-Curtis distance algorithm and the Anosim function, was employed to examine the similarity in microbial community structure between samples45,46,47. To assess the relationships among fungal genera, we employed Spearman’s rank correlation based on their relative abundances. To predict the ecological guilds of the fungal community, we used FUNGuild (version 1.0) following the guidelines described by Nguyen et al.48. Multiple- and two-group comparisons were conducted to compare the means of all samples within each group. Differential abundance analysis of microbial communities was performed using ALDEx249,50,51. The Kruskal-Wallis H test and Wilcoxon rank-sum test for differential abundance analysis were performed using the Majorbio Cloud platform.
Results
Sequencing results and microbial diversity
A total of 508,514 bacterial and 467,371 fungal sequences were obtained by paired-end sequence quality control and assembly (Tables S1 and S2). After rarefaction of sequence depth, bacterial community coverage ranged from 97.62 to 98.23%, and fungal community coverage ranged from 99.60 to 99.90% across all soil samples (Tables S3 and S4). The effective sequence counts and corresponding OTU numbers for each sample are listed in Tables S5 and S6. The results indicated that most bacterial and fungal taxa were detected, and that the sequencing depth was sufficient to capture the overall microbial composition across all soil samples.
Compared with those of the CK group, the Chao and Shannon indices of the bacterial community exhibited a decline in the Hh groups (Hh1 and Hh2), with the Hh2 group displaying the lowest values (Fig. 3a and b). Conversely, the Chao diversity index of the fungal community exhibited an increase in the Hh groups compared with the CK group, whereas the Shannon diversity index decreased, reaching its lowest value in the Hh2 group (Fig. 3c and d). These results suggest that H. hiemale alters the evenness of the fungal and bacterial communities and reduces the diversity of the bacterial community.

Alpha diversity of bacterial and fungal communities among the three groups. The Chao (a) and Shannon (b) indices of the bacterial community; The Chao (c) and Shannon (d) indices of the fungal community. Asterisks indicate statistical significance levels between pairwise comparisons: *** P < 0.001; ** P < 0.01; * P < 0.05.
Bacterial community composition
At the OTU level, the three groups shared 3,077 OTUs, with the Hh1 group having the highest number of unique OTUs (696) and the CK group having the lowest (533) (Fig. 4a). The Hh groups shared the highest number of OTUs (800). Pseudomonadota and Actinobacteriota were the dominant phyla in all three groups (Fig. S1). Acidobacteriota was less abundant in the Hh groups than in the CK group.
A ternary diagram reveals the distribution proportions of the dominant bacterial genera across the three groups (Fig. 4b). Notably, Nostoc_PCC-7524, Sphingomonas, and Bacillus spp. exhibited elevated distribution proportions in the Hh groups than in the CK group. Specifically, Bacillus displayed the highest distribution proportion in the Hh2 group, whereas Nostoc_PCC-7524 was predominant in the Hh1 group. In the CK group, the proportions of dominant bacterial genera such as RB41 was higher. Other genera showed similar proportional distribution across all three groups.

Bacterial community composition among three groups. (a) Venn diagram of bacterial OTU number; (b) The ternary diagram of dominant bacterial genera on genus level.
Fungal community composition
Overall, the three groups shared 529 OTUs. The CK group had the highest number of unique OTUs (242), whereas the Hh2 group had the lowest (126) (Fig. 5a). The Hh groups shared the highest number of OTUs (305). Ascomycota and Basidiomycota were the dominant phyla across the three groups (Fig. S2).
The ternary diagram displays the distribution proportions of dominant fungal genera in the three groups (Fig. 5b). Peziza, Hebeloma, g__unclassified_p__Rozellomycota, and g__unclassified_k__Fungi were present in higher proportions in the Hh groups than in the CK group. Hebeloma was the most abundant in the Hh2 group, and was not present in the CK group. unclassified_f__Thelephoraceae, Penicillium, Mortierella, Fusarium, Chaetomium, and Talaromyces were present in higher proportions in the CK group, with unclassified_f__Thelephoraceae being unique to this group.

Fungal community composition of the three groups. (a) Venn diagram of fungal OTU number; (b) The ternary diagram of dominant fungal genera.
Differences in structure and species in microbial communities
Variations in the bacterial and fungal communities among the three groups were analyzed using NMDS (Fig. 6). Notably, the bacterial community structure displayed greater divergence between the different groups compared with the fungal community structure. This indicated that the structure of the bacterial and fungal communities was altered by H. hiemale.
Analysis of the average relative abundance among the three groups revealed significant variations in the fungal and bacterial communities at the genus level (Tables S7 and S8). In the fungal community, Hebeloma was more abundant in the Hh2 group than in the other groups, Paraphoma was more abundant in the Hh1 group. In the bacterial community, Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium, Phenylobacterium, Massilia, and Burkholderia-Caballeronia-Paraburkholderia were more abundant in the Hh2 group than in the other groups; MND1 was more abundant in the CK group.

Differences in microbial community structure among three groups. NMDS analysis of bacterial (a) and fungal (b) communities at OTU level.
Species correlation analysis
Fungal correlation analysis at the genus level revealed that both Hebeloma and Peziza were negatively correlated with the majority of fungi, including Penicillium and Trichoderma (Fig. 7). Although the fungal correlation analysis did not show a significant positive correlation between Hebeloma and Peziza, a comparison of their abundance across the three groups indicated an increase in Peziza following the presence of Hebeloma (Fig. 5b).

Fungal correlation heatmap analysis at genus level. Asterisks denote statistical significance levels: *** P < 0.001; ** P < 0.01; * P < 0.05.
Functional predictions
FUNGuild was used to predict the functional profiles of fungal communities (Fig. S3). The results showed that ectomycorrhizae, represented by H. hiemale, and endophytes were significantly more abundant in the Hh2 group than in the CK group. Conversely, plant pathogen decreased when H. hiemale abundance increased.
Functional predictions of bacterial communities were performed using PICRUSt2, BugBase, and FAPROTAX. Notably, PICRUSt2 functional predictions revealed an increase in the abundance of H. hiemale resulted in an increase in the enzyme-level abundance of Monosaccharide-transporting ATPase and Serine-type D-Ala-D-Ala carboxypeptidase (Fig. S4), as well as the glyoxylate cycle and superpathway of the glyoxylate bypass-TCA in the MetaCyc pathway (Fig. S5), with the highest abundance observed in the Hh2 group. BugBase analysis revealed that the abundance of Contains Mobile Elements traits was significantly higher in the Hh groups than in the CK group (Fig. S6). In contrast, Potentially Pathogenic bacteria were significantly lower in the Hh groups than in the CK group, and Anaerobic bacteria were significantly lower in the Hh2 group than in the CK group. FAPROTAX functional predictions indicated that following H. hiemale inoculation, functions related to nitrogen fixation and photoautotrophy were enhanced, with a higher abundance in the Hh groups than in the Ck group (Fig. S7).
Discussion
Analysis of microbial diversity and community structure
Microbial diversity analyses revealed that inoculation with H. hiemale significantly affected both the diversity and evenness of the fungal and bacterial communities (Fig. 3). The bacterial Shannon and Chao indices decreased in the Hh1 and Hh2 groups compared with the CK group, with Hh2 showing the lowest values. In contrast, although the fungal Chao index increased with H. hiemale inoculation, the fungal Shannon index decreased, reaching its lowest value in the Hh2 group. NMDS analysis revealed distinct clustering patterns among the three groups (Fig. 6), suggesting that H. hiemale inoculation differentially influenced the structure of the bacterial and fungal communities. The bacterial community appeared to diverge more than the fungal community did after inoculation with H. hiemale. These results suggest that H. hiemale may favor certain microbes in bacterial and fungal communities, thereby reducing the overall bacterial diversity while reshaping fungal evenness. Similar observations of mycorrhizal fungi altering rhizosphere diversity have been reported in other studies52,53. The reduced bacterial diversity suggests competitive exclusion of some microbes by those that are better adapted to the ectomycorrhizal environment54,55. A possible explanation for this is that ECM can release exudates or modify root exudation patterns that selectively promote or suppress certain microbes8,56.
Analysis of microbial community composition
Analysis of microbial bacterial community composition
In the present study, we observed significant differences in the abundances of several bacterial genera among the three groups. These findings suggest that the presence of H. hiemale can significantly influence the composition of bacterial communities in the rhizosphere of host plants. Previous research has shown that ECM symbionts can selectively affect associated bacterial communities55. Our results further support this finding, indicating that H. hiemale modulates the rhizosphere microbiome by promoting the growth of specific microbial communities.
Notably, the abundances of Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium, Massilia, Burkholderia-Caballeronia-Paraburkholderia, Luteibacter, Dyella, Chitinophaga were significantly higher in the CK group than in the Hh2 group. Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium is known for its nitrogen-fixing capabilities, which play crucial roles in enhancing host plant growth by improving nitrogen availability57,58. Massilia has been identified as possessing phosphorus-solubilization abilities59 which can facilitate the absorption of available phosphorus by the host plant, thereby promoting host plant growth. Additionally, a species of the genus Massilia has been found to produce dimethyl disulfide, offering a potential alternative soil fumigant to replace methyl bromide60, which could help mitigate the adverse effects of soil-borne diseases and nematodes. Burkholderia-Caballeronia-Paraburkholderia plays a significant role in degrading atrazine, a pesticide residue, showing a strong positive correlation with the rate of atrazine degradation61. Luteibacter, which is associated with cellulose degradation, contributes to enhancement of soil fertility62, and has also been shown to degrade methamidophos, another pesticide residue63. Dyella participates in the selenium cycle by converting organic selenium into selenium-oxygen anions, which facilitate selenium uptake by the host plant64,65. Chitinophaga has emerged as a potential key player in suppressing significant plant pathogens, such as Ralstonia solanacearum, contributing to the healthy growth of the host plant66. Their diverse ecological roles, such as nitrogen fixation, and pollutant degradation, contribute to the maintenance of soil fertility and promotion of sustainable agricultural practices. Notably, Luteibacter was exclusively detected in the Hh groups and was absent in the CK group. Thus, H. hiemale may indirectly contribute to improved host plant health and growth by selectively shaping bacterial community composition.
Analysis of microbial fungal community composition
Inoculation with H. hiemale altered the fungal community composition in the rhizosphere of the host plant, significantly increasing the abundance of unclassified_p__Mortierellomycota and Lecythophora. A notable increase in their relative abundance was observed in the CK group compared to that in the Hh2 group. Interestingly, Mortierellomycota has been reported to exhibit a significant negative correlation with biogenic Cd levels67 and may represent a key component of the rhizosphere microbiome that enhances the resistance of alfalfa to Cd68. This suggests that the increased abundance of unclassified_p__Mortierellomycota, induced by H. hiemale inoculation, may contribute to the mitigation of Cd toxicity in Q. mongolica. Furthermore, the increased abundance of Lecythophora in response to H. hiemale inoculation may have environmental implications. Lecythophora exhibits significant resistance to Hg(II) and can volatilize Hg(II) to Hg(0), potentially improving mercury-contaminated environments69,70. Lecythophora abundance increased with H. hiemale inoculation, which could potentially enhance this mercury-remediation process, contributing to the overall health of the ecosystem. In addition, the ternary diagram (Fig. 5) revealed that Peziza abundance was higher in the Hh groups than in the CK group. The fungal correlation heatmap analysis (Fig. 7) also indicated a positive correlation between Peziza and Hebeloma. This observation aligns with previous findings that Peziza frequently co-occurs with a specific ECM, suggesting potential facilitative interactions or similar nutrient requirements71,72.
In contrast to its positive effects on unclassified_p__Mortierellomycota and Lecythophora, H. hiemale inoculation appeared to inhibit the abundance of Fusarium. As the concentration of H. hiemale increased, a corresponding decrease in the relative abundance of Fusarium was observed. This suggests that H. hiemale possesses certain compounds or properties that selectively suppress the growth of Fusarium. Given the pathogenic nature of some species within this genera73,74,75, the inhibitory effect of H. hiemale may provide an additional layer of protection for the host plant, promoting its overall health and resilience. Inoculation with H. hiemale influenced the fungal community composition in the rhizosphere of the host plant, with notable increases in the abundance of unclassified_p__Mortierellomycota and Lecythophora, and a concomitant decrease in Fusarium. These changes in fungal community structure may have important implications for the host plant, including reduced Cd uptake, enhanced Hg remediation, and increased protection against potential pathogens.
Functional predictive analysis of microbial communities
Functional prediction analysis of fungal community
FUNGuild predicted that functional analysis of the fungal community would reveal significant changes after H. hiemale inoculation. The results demonstrated that ectomycorrhizal and endophytic fungi were significantly increased in the Hh2 group compared with the CK group. Previous studies have shown that the community structures of ectomycorrhizal and endophytic fungi can influence one another through interspecific competitive and facilitative interactions in plant roots76,77. Therefore, the observed increase in endophytes suggests the potential for facilitative interactions between H. hiemale and endophytes.
Conversely, FUNGuild predicted that the plant pathogens would decrease with H. hiemale abundance increased. For example, within the fungal community, the presence of H. hiemale also leads to a significant decrease in the abundance of Fusarium, a genus of filamentous fungi that includes many plant pathogens73,74,75. H. hiemale may indirectly promote the growth and health of host plants by suppressing the growth of plant pathogens. The observed changes in fungal community composition and function following H. hiemale inoculation highlight the complex interactions among ECM, microbial community, and host plants. This suggests a potential mechanism by which the ECM can shape the microbiome to benefit the host plant.
Functional prediction analysis of bacterial community
In this study, we employed a multifaceted approach to investigate the effect of ectomycorrhiza on the functional profiles of bacterial communities. PICRUSt2 showed that the enzyme-level abundance of monosaccharide-transporting ATPase increased with the increasing abundance of H. hiemale, with the highest abundance observed in the Hh2 group. Furthermore, MetaCyc pathway analysis revealed an increase in the abundance of the superpathway of glyoxylate bypass-TCA and glyoxylate cycle. This phenomenon may be attributed to the competitive dynamics established between H. hiemale and the bacterial community. The competition for carbon sources between H. hiemale and the bacterial community likely drove the elevation of the enzyme-level abundance of monosaccharide-transporting ATPase within the bacterial community, as it is critical for accelerated carbon metabolism78. Moreover, competition might have resulted in carbon source deficiency, consequently stimulating biomolecule synthesis by upregulating the superpathway of glyoxylate bypass-TCA and glyoxylate cycle, which are essential pathways for the synthesis of biomolecules under carbon-limiting conditions79.
BugBase prediction analysis showed that the abundance of Containing Mobile Elements was higher in the Hh groups than in the CK group. Conversely, Anaerobic was significantly lower in the Hh2 group than in the CK group. Mobile elements, such as transposons and plasmids, facilitate horizontal gene transfer in bacteria, which can profoundly impact their fitness and evolution80,81. The observed increase in Containing Mobile Elements may indicate an adaptive response of the bacterial community to the presence of H. hiemale. Mycorrhizal activity can influence the soil pore spaces available for microbial habitation, resulting in altered oxygen concentrations82. The observed decrease in anaerobes suggests that H. hiemale inoculation may modify oxygen availability in the rhizosphere, thereby potentially suppressing the growth of anaerobic bacteria. Predictions generated by the FAPROTAX function indicated elevated levels of nitrogen fixation with increasing H. hiemale inoculation. Nitrogen fixation activity may indicate enhanced nitrogen uptake by the host plant upon inoculation with H. hiemale, as this process is crucial for the conversion of atmospheric nitrogen into bioavailable forms for plant uptake83,84.
Conclusion
In this study, diversity and composition of microbial communities in the rhizosphere of the host plant were compared after inoculation with H. hiemale. Compared with the CK group, H. hiemale inoculation altered the evenness of both fungal and bacterial communities and reduced the diversity of the bacterial community. Notable changes were observed in the bacterial community composition, with several genera exhibiting an enhanced capacity for nutrient cycling, pollutant degradation, and host plant protection. Shifts in the fungal community structure may have significant implications for the host plant, such as reduced Cd uptake, enhanced mercury remediation, and increased protection against potential pathogens. It provides valuable insights into the intricate interactions between ECM and microbial communities in the rhizosphere, underscoring the need to account for multi-species interactions to understand the ecological implications of plant–microbe relationships. Further research is necessary to elucidate the specific mechanisms underlying these effects and their potential impact on host plant fitness. Future studies should explore the potential applications of H. hiemale in sustainable agriculture and environmental management, which could lead to novel strategies for increasing crop productivity and mitigating environmental pollution.
Data availability
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: National Center for Biotechnology Information (NCBI) BioProject (PRJNA1116842).
References
Allen, M. F., Swenson, W., Querejeta, J. I., Egerton-Warburton, L. M. & Treseder, K. K. Ecology of mycorrhizae: a conceptual framework for complex interactions among plants and fungi. Annu. Rev. Phytopathol. 41, 271–303 (2003).
Genre, A., Lanfranco, L., Perotto, S. & Bonfante, P. Unique and common traits in mycorrhizal symbioses. Nat. Rev. Microbiol. 18, 649–660 (2020).
Pohjanen, J. et al. Interaction with ectomycorrhizal fungi and endophytic Methylobacterium affects nutrient uptake and growth of pine seedlings in vitro. Tree Physiol. 34, 993–1005 (2014).
Seb, J., Ajungla, T., ROLE OF ECTOMYCORRHIZA & IN FOREST ECOSYSTEMS: A REVIEW. IJAR 6, 866–873 (2018).
Shi, J., Wang, X. & Wang, E. Mycorrhizal symbiosis in plant growth and stress adaptation: from genes to ecosystems. Annu. Rev. Plant. Biol. 74, 569–607 (2023).
Gay, G. Effect of the ectomycorrhizal fungus Hebeloma hiemale on adventitious root formation in derooted Pinus halepensis shoot hypocotyls. Can. J. Bot. 68, 1265–1270 (1990).
Elbashiti, T., El Kichaoui, A. & Ajwa, A. Evaluation the effect of ectomycorrhizal fungi on Prunus cerasifera X salicina (Rosaceae) growth compared with chemical and organic fertilizer. Sky J. Agricultural Res. 6, 41–49 (2017).
Courty, P. E. et al. The role of ectomycorrhizal communities in forest ecosystem processes: new perspectives and emerging concepts. Soil Biol. Biochem. 42, 679–698 (2010).
Liu, Y., Li, X. & Kou, Y. Ectomycorrhizal fungi: participation in nutrient turnover and community assembly pattern in forest ecosystems. Forests 11, 453 (2020).
Zhang, Y. et al. Lycoperdon perlatum from a coniferous forest forms an ectomycorrhizal relation with and increases drought resistance of Populus × Canadensis ‘zhongliao 1’. Symbiosis https://doi.org/10.1007/s13199-025-01035-4 (2025).
Stuart, E. K. et al. Mycocentric fertilisation of ectomycorrhizae-inoculated Pinus radiata during containerised production alters root Microbiome and growth outcomes. Australian Forestry. 86, 110–128 (2023).
Martínez de Aragón, J. et al. Economically profitable post fire restoration with black truffle (Tuber melanosporum) producing plantations. New Forest. 43, 615–630 (2012).
Keyi, C., Huiru, Z. & Xiangdong, L. Spatial pattern of Quercus mongolica in natural secondary forest. Acta Ecol. Sinica 38, (2018).
Han, X., Liang, X., Ma, S., Wang, Y. & Lu, X. Depletion of stem non-structural carbohydrates supports release of lateral branches from apical control in Quercus mongolica seedlings. Trees 36, 1573–1584 (2022).
Bai, X. N., Hao, H., Hu, Z. H. & Leng, P. S. Ectomycorrhizal inoculation enhances the salt tolerance of Quercus mongolica seedlings. Plants (Basel). 10, 1790 (2021).
Castrillo, G. et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature 543, 513–518 (2017).
Kwak, M. J. et al. Rhizosphere Microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. https://doi.org/10.1038/nbt.4232 (2018).
Shelake, R. M., Pramanik, D. & Kim, J. Y. Exploration of Plant-Microbe interactions for sustainable agriculture in CRISPR era. Microorganisms 7, 269 (2019).
Rodriguez, P. A. et al. Systems biology of Plant-Microbiome interactions. Mol. Plant. 12, 804–821 (2019).
Nadarajah, K. & Abdul Rahman, N. S. N. Plant–Microbe interaction: aboveground to belowground, from the good to the bad. Int. J. Mol. Sci. 22, 10388 (2021).
Haney, C. H., Samuel, B. S., Bush, J. & Ausubel, F. M. Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nat. Plants. 1, 1–9 (2015).
Mendes, R. et al. Deciphering the rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 332, 1097–1100 (2011).
Shabuer, G. et al. Plant pathogenic anaerobic bacteria use aromatic polyketides to access aerobic territory. Science 350, 670–674 (2015).
Doehlemann, G., Ökmen, B., Zhu, W. & Sharon, A. Plant pathogenic Fungi. Microbiol Spectr 5, (2017).
Xu, G. et al. A fungal effector targets a heat shock–dynamin protein complex to modulate mitochondrial dynamics and reduce plant immunity. Sci. Adv. 6, eabb7719 (2020).
Edwards, J. D. et al. Soil microbial community response to ectomycorrhizal dominance in diverse Neotropical montane forests. Mycorrhiza https://doi.org/10.1007/s00572-023-01134-4 (2024).
Berrios, L. et al. Ectomycorrhizal fungi alter soil food webs and the functional potential of bacterial communities. mSystems 0, e00369–e00324 (2024).
Guo, H. et al. Soil Microbiome of Shiro reveals the symbiotic relationship between tricholoma Bakamatsutake and Quercus mongolica. Front Microbiol 15, (2024).
Li, Q. et al. Tuber indicum shapes the microbial communities of ectomycorhizosphere soil and ectomycorrhizae of an Indigenous tree (Pinus armandii). PLOS ONE. 12, e0175720 (2017).
Li, Q. et al. Chinese black truffle (Tuber indicum) alters the ectomycorrhizosphere and endoectomycosphere Microbiome and metabolic profiles of the host tree Quercus Aliena. Frontiers Microbiology 9, (2018).
Edwards, J. et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. 112, E911–E920 (2015).
Al-Askar, A. & Rashad, Y. Arbuscular mycorrhizal fungi: A biocontrol agent against common bean fusarium root rot disease. Plant Pathol. Journal 9, (2010).
Jia, T. Characterization of cucumber rhizosphere bacterial community with high-throughput amplicon sequencing. AJ 47, 103–112 (2019).
Fadiji, A. E., Kanu, J. O. & Babalola, O. O. Impact of cropping systems on the functional diversity of rhizosphere microbial communities associated with maize plant: a shotgun approach. Arch. Microbiol. 203, 3605–3613 (2021).
You, Y. et al. Contrasting mechanisms of Poplar and rhizosphere soil influenced by Urea and compound fertilizer. Ind. Crops Prod. 229, 120995 (2025).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 10, 996–998 (2013).
STACKEBRANDT, E., GOEBEL, B. M. & Taxonomic Note A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. MicroBiol. 44, 846–849 (1994).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Abarenkov, K. et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: sequences, taxa and classifications reconsidered. Nucleic Acids Res. 52, D791–D797 (2024).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688 (2020).
Schloss, P. D. et al. Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Chapman, M. G. & Underwood, A. J. Ecological patterns in multivariate assemblages: information and interpretation of negative values in ANOSIM tests. Mar. Ecol. Prog. Ser. 180, 257–265 (1999).
Bray, J. R. & Curtis, J. T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 27, 326–349 (1957).
Kruskal, J. B. Nonmetric multidimensional scaling: A numerical method. Psychometrika 29, 115–129 (1964).
Nguyen, N. H. et al. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248 (2016).
Fernandes, A. D., Macklaim, J. M., Linn, T. G., Reid, G. & Gloor, G. B. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS One. 8, e67019 (2013).
Fernandes, A. D. et al. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2, 15 (2014).
Nearing, J. T. et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 13, 342 (2022).
Alhuthali, S. et al. Soil physicochemical and metagenomic analyses of Bacteria and fungi: toward desert truffle cultivation in Saudi Arabia. Agronomy 14, 3021 (2024).
Li, J. et al. Arbuscular mycorrhizal fungi enhance drought resistance and alter microbial communities in maize rhizosphere soil. Environ. Technol. Innov. 37, 103947 (2025).
Guo, X., Wan, Y., Shakeel, M., Wang, D. & Xiao, L. Effect of mycorrhizal fungi inoculation on bacterial diversity, community structure and fruit yield of blueberry. Rhizosphere 19, 100360 (2021).
Izumi, H. & Finlay, R. D. Ectomycorrhizal roots select distinctive bacterial and ascomycete communities in Swedish Subarctic forests. Environ. Microbiol. 13, 819–830 (2011).
Yang, Q. et al. Influence of Mikania micrantha Kunth flavonoids on composition of soil microbial community. Int. J. Mol. Sci. 26, 64 (2025).
Zhou, Y. et al. Sex-based metabolic and microbiota differences in roots and rhizosphere soils of dioecious Papaya (Carica Papaya L). Front. Plant. Sci. 13, 991114 (2022).
Mousavi, S. A. et al. Phylogeny of the Rhizobium-Allorhizobium-Agrobacterium clade supports the delineation of Neorhizobium gen. Nov. Syst. Appl. Microbiol. 37, 208–215 (2014).
Zheng, B. X., Bi, Q. F., Hao, X. L., Zhou, G. W. & Yang, X. R. Massilia phosphatilytica Sp. nov., a phosphate solubilizing bacteria isolated from a long-term fertilized soil. Int. J. Syst. Evol. Microbiol. 67, 2514–2519 (2017).
Feng, G. D., Yang, S. Z., Li, H. P. & Zhu, H. H. Massilia Putida Sp. nov., a dimethyl disulfide-producing bacterium isolated from Wolfram mine tailing. Int. J. Syst. Evol. Microbiol. 66, 50–55 (2016).
Zhao, Y. et al. Rapid biodegradation of atrazine by a novel paenarthrobacter ureafaciens ZY and its effects on soil native microbial community dynamic. Front. Microbiol. 13, 1103168 (2022).
López-Mondéjar, R., Zühlke, D., Becher, D., Riedel, K. & Baldrian, P. Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci. Rep. 6, 25279 (2016).
Wang, L., Wang, G., Li, S. & Jiang, J. Luteibacter jiangsuensis Sp. nov.: a methamidophos-degrading bacterium isolated from a methamidophos-manufacturing factory. Curr. Microbiol. 62, 289–295 (2011).
Wang, Y. et al. Linking microbial community composition to farming pattern in selenium-enriched region: potential role of microorganisms on se geochemistry. J. Environ. Sci. (China). 112, 269–279 (2022).
Luo, X. et al. Microbial oxidation of organic and elemental selenium to selenite. Sci. Total Environ. 833, 155203 (2022).
Deng, X. et al. Soil Microbiome manipulation triggers direct and possible indirect suppression against Ralstonia solanacearum and fusarium oxysporum. NPJ Biofilms Microbiomes. 7, 33 (2021).
Shi, Y. et al. K fertilizers reduce the accumulation of cd in Panax Notoginseng (Burk.) F.H. By improving the quality of the microbial community. Front. Plant. Sci. 11, 888 (2020).
Wang, X. et al. Improvement of alfalfa resistance against cd stress through rhizobia and arbuscular mycorrhiza fungi co-inoculation in cd-contaminated soil. Environ. Pollut. 277, 116758 (2021).
Chang, J. et al. Bioremediation of Hg-contaminated soil by combining a novel Hg-volatilizing lecythophora Sp. fungus, DC-F1, with Biochar: performance and the response of soil fungal community. Sci. Total Environ. 671, 676–684 (2019).
Chang, J. et al. Transcriptomic analyses reveal the pathways associated with the volatilization and resistance of mercury(II) in the fungus lecythophora Sp. DC-F1. Sci. Total Environ. 752, 142172 (2021).
Hrynkiewicz, K., Szymańska, S., Piernik, A. & Thiem, D. Ectomycorrhizal community structure of Salix and Betula spp. At a saline site in central Poland in relation to the seasons and soil parameters. Water Air Soil. Pollut. 226, 99 (2015).
Zhong, F. et al. Soil fungal community composition and diversity of culturable endophytic Fungi from plant roots in the reclaimed area of the Eastern Coast of China. J. Fungi. 8, 124 (2022).
Ma, L. J. et al. Fusarium pathogenomics. Annu. Rev. Microbiol. 67, 399–416 (2013).
Summerell, B. A. Resolving fusarium: current status of the genus. Annu. Rev. Phytopathol. 57, 323–339 (2019).
Perincherry, L., Lalak-Kańczugowska, J. & Stępień, Ł. Fusarium-Produced Mycotoxins in Plant-Pathogen interactions. Toxins (Basel). 11, 664 (2019).
Yamamoto, S. et al. Spatial segregation and aggregation of ectomycorrhizal and Root-Endophytic Fungi in the seedlings of two Quercus species. PLOS ONE. 9, e96363 (2014).
Toju, H., Yamamoto, S., Tanabe, A. S., Hayakawa, T. & Ishii, H. S. Network modules and hubs in plant-root fungal biomes. J. R. Soc. Interface. 13, 20151097 (2016).
Xu, W. et al. Dibutyl phthalate alters the metabolic pathways of microbes in black soils. Sci. Rep. 8, 2605 (2018).
Lorenz, M. C. & Fink, G. R. Life and death in a macrophage: role of the glyoxylate cycle in virulence. Eukaryot. Cell. 1, 657–662 (2002).
Arber, W. Horizontal gene transfer among Bacteria and its role in biological evolution. Life (Basel). 4, 217–224 (2014).
Vale, F. F., Lehours, P., Yamaoka, Y. & Editorial The role of mobile genetic elements in bacterial evolution and their adaptability. Front. Microbiol. 13, 849667 (2022).
Rillig, M. C. & Mummey, D. L. Mycorrhizas and soil structure. New. Phytol. 171, 41–53 (2006).
Zhang, L. et al. A highly conserved core bacterial microbiota with nitrogen-fixation capacity inhabits the xylem Sap in maize plants. Nat. Commun. 13, 3361 (2022).
Guo, K., Yang, J., Yu, N., Luo, L. & Wang, E. Biological nitrogen fixation in cereal crops: progress, strategies, and perspectives. Plant. Commun. 4, 100499 (2023).
Funding
This work was supported by the Natural Science Foundation of Liaoning Province (2024-MSLH-344), the National Natural Science Foundation of China (32370008), the Natural Science Foundation of Liaoning Province(2022-MS-418), and Research and Demonstration of Edible and Medicinal Fungi Industry in Nielamu County - Development Plan of Xizang (XZ202201ZY0019N).
Author information
Authors and Affiliations
Contributions
HB-G: Writing – original draft, review & editing, Methodology, Formal analysis. JC-Z: Writing – original draft, Methodology, Software, Formal analysis. WY-L: Writing – original draft, Investigation, Formal analysis, Data curation. YD-B: Writing – original draft, Visualization, Conceptualization. LA-S: Writing – review & editing, Visualization, Conceptualization. XD-Y: Writing – review & editing, Supervision, Conceptualization.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, HB., Zhao, JC., Liu, WY. et al. Microbiome analysis for artificially establishing the symbiotic relationship between Hebeloma hiemale and Quercus mongolica. Sci Rep 15, 19273 (2025). https://doi.org/10.1038/s41598-025-03963-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03963-z
