
Abstract
SARS-CoV-2, the virus responsible for COVID-19, exhibits structural differences compared to other coronaviruses that impact transmission dynamics and host response. Although vaccines have reduced severe hospitalizations and deaths, understanding the infection remains challenging, and finding viable biomarkers for disease severity would be beneficial. In this context, microRNAs (miRNAs) are a set of small non-coding RNAs that have emerged as potential biomarkers for numerous diseases. However, assessing miRNA expression requires the normalization of RT-qPCR data using appropriate endogenous reference genes. The selection of these reference genes remains a major challenge, directly impacting the accuracy of expression analyses. Although several small RNAs, including snRNAs, have been evaluated, there is still no consensus regarding the optimal reference genes for the normalization of plasma miRNA expression data in COVID-19 infection. This study evaluated five candidate genes—including RNU6B, snRNA U6 (small nuclear RNAs) and three miRNAs (miR-320a, miR-342-3p, and miR-328) as potential endogenous normalizers for analyzing miRNA expression in the plasma of COVID-19 patients. The snRNA U6 showed greater stability using a combination of statistical algorithms (NormFinder, RefFinder, BestKeeper, and GeNorm), indicating that this snRNA can be used as a robust internal reference for analysis in the plasma of COVID-19 patients.
Similar content being viewed by others

Introduction
The coronavirus disease 2019 (COVID-19), caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), has resulted in a pandemic that necessitated significant global efforts to understand the virus and the clinical course of the disease1. Most infected individuals exhibit mild clinical manifestations resembling those of a cold, including symptoms such as fever, cough, malaise, myalgia, headache, and disturbances in taste and smell. However, some individuals may develop severe pneumonia and acute respiratory distress syndrome (ARDS)1,2.
The pattern of clinical manifestations has evolved throughout the pandemic, primarily due to two factors: (1) the development of vaccines against COVID-19 and (2) the emergence of new variants and subvariants. Furthermore, socio-economic disparities and sanitation inequalities have been associated with worse health outcomes3,4. To mitigate the public health impact of COVID-19, coordinated global actions were essential, including the implementation of non-pharmaceutical interventions and vaccine development.
In Brazil, vaccination began in January 2021, leading to a significant reduction in mortality. Protection against death decreased by 50% within 20 weeks after the initial vaccination series, and protection against severe disease declined by 25% after 19 weeks5. Despite the introduction of vaccines and the consequent increase in vaccination coverage, SARS-CoV-2 remains a global concern due to the multiple waves of COVID-19 that have developed during the pandemic, driven by the emergence of new variants. These variants have been a key factor contributing to the occurrence of these waves6. Furthermore, individual-related factors are crucial for disease outcomes, making it essential to understand the underlying mechanisms of the infection and their implications for host response and long-term consequences. In this context, microRNAs (miRNAs) may be critical for elucidating the diverse clinical manifestations of COVID-19 and hold potential as biomarkers for disease progression and severity7.
In this context, small non-coding RNAs, including microRNAs (miRNAs) and small nuclear RNAs (snRNAs), may be critical for elucidating the diverse clinical manifestations of COVID-19 and hold potential as biomarkers for disease progression and severity. Accurate detection and quantification of miRNAs in biological fluids, such as plasma, provide insights that enhance our understanding of the dynamics of infection and host response. These short (~ 18–25 nucleotides), single-stranded molecules regulate gene expression post-transcriptionally and may directly influence immune and inflammatory responses to infectious diseases like COVID-198. Released into the extracellular space, miRNAs are encapsulated in macrovesicles or exosomes or bound to high-density lipoproteins or protein complexes9. Thus, these molecules can develop stability in various body fluids, such as serum, plasma, saliva, urine, and milk, making them non-invasive biomarkers, constituting promising for molecular diagnoses and prognostics10,11. This potential has been reported across various diseases12,13, including cancer, diabetes14,15, cardiovascular diseases16, and COVID-1917.
Although the stability of miRNAs in body fluids is measurable, the precise quantification of miRNA levels remains essential due to the influence of multiple external factors. The quality and quantity of the analyzed material may be susceptible to interferences related to collection, storage, RNA extraction processes, as well as the performance of reverse transcriptase (RT) and polymerase chain reaction (PCR). Such discrepancies can introduce bias in miRNA quantification, compromising their use as molecular markers and affecting the evaluation of differentially expressed miRNA functions. Thus, accuracy and consistency in miRNA quantification are fundamental for obtaining reliable results in studies utilizing these molecular markers18,19.
The interpretation of miRNA expression data requires appropriate normalization using endogenous reference genes, a critical step that can significantly influence the results. In this context, the selection of suitable reference genes remains a challenge, particularly in plasma samples from COVID-19 patients. This study aims to evaluate the stability of five small RNAs (snRNAs and miRNAs) as potential endogenous normalizers, contributing to the validation of robust analytical methods for expression studies in clinical contexts.
Results
Selection of candidate reference small RNAs (snRNAs and miRNAs) and stability analysis
The study population consisted of 48 individuals, with a mean age of 39.02 years (median = 36.0), ranging from 18 to 76 years. Sex distribution showed a higher prevalence of female participants (29; 60.42%) compared to male participants (19; 39.58%). Regarding clinical status, most participants did not require hospitalization (45; 93.75%), while 3 (6.25%) were hospitalized (Table 1). The initial selection of candidate small RNAs and their stability analysis were performed by evaluating the expression of five candidate genes—two small nuclear RNA (RNU6B and snRNA U6) and three miRNAs (miR-320a, miR-342-3p, and miR-328) in plasma samples from the case (n = 24) and control (n = 24) groups. However, one control sample was excluded from the final analysis due to insufficient plasma volume collected. Although we acknowledge the limitation of our sample size, we chose to focus exclusively on COVID-19 patients and viral-negative controls to ensure a homogeneous population that reflects the unique molecular alterations associated with SARS-CoV-2 infection. Including samples from other respiratory diseases, while potentially enhancing generalizability, was beyond the scope of this study.
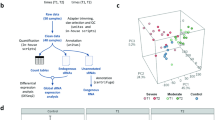
Expression analysis was based on the quantitative cycle, evaluating the mean cycle threshold (Ct). All datasets of input RNA and inter-run calibrator-normalized Ct-values were analyzed with the NormFinder software to perform stability analysis, estimating the values for intra- and inter-group variability. RNU6B, a small nuclear RNA (snRNA) commonly used as a normalizer for intracellular and circulating miRNA expression analyses, exhibited low expression levels and poor homogeneity among plasma samples. Its amplification was undetectable in 95.8% of the plasma samples tested in duplicate, indicating very low and inconsistent expression levels across plasma samples (data no show). Besides, these analysis also indicated that, among the candidate genes, snRNA U6 presented the most stable expression (Stability value = 0.298) (Fig. 1).

Analysis of endogenous normalizers stability using NormFinder algorithm.
Additionally, RefFinder software was used to analyze the same data set. This tool conjugates the normalization determination algorithms GeNorm, BestKeeper, comparative ΔCt and NormFinder. Stability analysis conducted with BestKeeper revealed that snRNA U6 presented the most stable expression in the group (Fig. 2a)., followed by miR-320a, miR-328-3p and miR-342-3p, subsequently. Conversely, miR-342-3p displayed the highest stability value, suggesting that it is the least suitable normalizer among the candidates in this dataset (Fig. 2a). The comparative ΔCt method (Fig. 2b) revealed the same results as the NormFinder online version included in RefFinder (Fig. 2d) or the GeNorm analysis (Fig. 2c) that indicated snRNA U6 as the most stable normalization candidate, followed by miR-342-3p, miR-320a and miR-328-3p. Assembling all the algorithms together by calculating the mean rank for each of the 4 candidate internal normalizers snRNA U6, followed by miR-342-3p, miR-320a and miR-328-3p were the best (Fig. 2e).
Currently, RefFinder is the only web-based platform designed to compare and assess housekeeping genes as potential reference genes. This tool consolidates four commonly used computational methods (geNorm, NormFinder, BestKeeper, and the comparative ΔCt method) into an online resource for evaluating the stability and reliability of reference genes. The stability rankings from each method are used to assign appropriate weights to the genes, and the geometric mean of these weights is calculated to determine the final ranking. Additionally, users can choose to employ a single program or a combination of the four to rank candidate reference genes20.

Assessment of endogenous reference gene stability using the different algorithms: BestKeeper, ΔCt, GeNorm and NormFinder.
Expression differences between case and control groups
The data were analysed between the two groups to identify any differences in mean Ct values, given the importance of ensuring that an internal reference gene exhibits similar expression levels in diseased and healthy conditions. In this context, no significant differences were observed between the case and control groups in the mean expression of snRNA U6, -328 and miR-342-3p (all p-value > 0.05). However, a significant difference was identified between the two groups in the expression of miR-320a (Fig. 3).

Comparative expression levels of normalization candidates in case individuals with SARS-CoV-2 RNA detectable by RT-qPCR and respiratory symptoms and control (individuals with SARS-CoV-2 RNA undetectable by RT-qPCR groups.
Discussion
The selection of a reliable internal reference gene (internal reference miRNA) is critical to ensure consistent expression across various health conditions and experimental groups. To achieve this, normalization using stable reference genes is an essential tool to eliminate technical variations and ensure that the observed changes in miRNA expression are biological rather than technical or artifactual21.
In this study, we analyzed potential candidate genes—two small nuclear RNAs (RNU6B and snRNA U6) and three miRNAs (miR-320a, miR-342-3p, and miR-328) as candidate reference genes for future differential expression studies in the context of the COVID-19 pandemic. We incorporated well-established statistical algorithms (comparative ΔCt method, geNorm, NormFinder, and BestKeeper) as tools for evaluating their stability.
Despite the widespread adoption of normalization strategies, there is no definitive guideline or universal recommendation for normalization in all experimental conditions18. Typically, endogenous control strategies rely on constitutively expressed miRNAs, either individually or as a combination of several miRNAs. Alternatively, normalization may employ the global mean Cq value from all analyzed miRNAs.
Initially, this study excluded RNU6B due to its low expression and homogeneity between samples, corroborating recent suggestions that it was not consistently expressed in plasma and serum samples22. RNU6B is small nuclear RNA (snRNA), frequently used as a reference in miRNA quantification, although its primary function is related to the modification and maturation of ribosomal RNAs, distinguishing between pathophysiological conditions. This miRNA has been commonly used in microRNA expression studies and has been employed as a reference normalizer in several investigations, including studies involving plasma samples from individuals co-infected with HIV-1 and HCV23, as well as COVID-19 patients24. However, in other investigations aimed at normalizing target miRNA expression in plasma from tuberculosis patients, RNU6B proved to be an unstable marker due to its high variability25,26. In fact, the low stability of RNU6B was corroborated in a more recent study, which demonstrated that it is not a stable marker in plasma samples, showing highly variable expression19,27.
Similarly, snRNA U6, another frequently used reference gene, has shown variability across studies. It functions as an essential component of the spliceosome, involved in the removal of introns during pre-mRNA processing. It has been employed in miRNA expression analyses in various contexts, including digestive malignancies (for example, hepatocellular carcinoma, gastric carcinoma, liver cirrhosis, and hepatitis B)28, as well as research into chronic kidney disease and nocturnal hypertension29. In this study, based on the quantitative cycle (Cq), no significant differences were observed between the case and control groups in relation to the average expression of snRNA U6 (p-value > 0.05). However, a study involving plasma from heart failure30 patients revealed that snRNA U6 exhibited high variability, rendering it unsuitable as an endogenous control in this setting. This variability suggests that miRNAs traditionally used for normalization may not be suitable under all conditions, indicating that their stability should be analyzed in different contexts21,31.
RNU6B and U6 are small nuclear RNAs (snRNAs) frequently employed as endogenous reference genes in miRNA expression studies, owing to their presumed constitutive expression and functional relevance in RNA processing. U6, in particular, is a core component of the spliceosome and plays an essential role in pre-mRNA splicing32,33,34. RNU6B, a transcript variant related to snRNA U6, has been widely used as a normalizer in RT-qPCR protocols. However, its expression has been shown to vary under different physiological and pathological conditions32. Moreover, studies on cancer and extracellular RNA have emphasized that even traditionally accepted reference RNAs such as RNU6B may not be appropriate for all experimental settings35. Recent studies, emphasize that these differences in their biogenesis and functions may lead to variations in stability and expression under different biological contexts. Such distinctions underpin the rationale for selecting these RNAs differentially for normalization in miRNA expression analyses, thereby enhancing the robustness of our results32,33,36,37.
In relation to miR-320a, significant difference in its expression was observed between the groups, suggesting instability. In fact, the miR-320a has been commonly used as a biomarker in various diseases due to its differential expression14,37,38,39, but not normally as a reference gene for miRNA normalization. In fact, miR-320a expression analysis has demonstrated certain variations in different contexts37,40,41. The data raises questions about the expression of miR-320a as potential differential marker in COVID-19 context.
The analysis of the average expression of miRNAs based on cycle threshold (Ct) values42 can be limiting, especially in studies comparing different health and disease conditions. Indeed, the exclusion of missing values, which is common in this analysis, can bias the results toward a favorable expression mean, disregarding the true heterogeneity of the data. This lack of significance may be attributed to variations in the concentrations of these miRNAs, which could be low, and these miRNAs may exhibit greater stability in different biological fluids.
To enhance the robustness of the analysis, the application of algorithms is a widely used technique, as it provides a more comprehensive assessment of gene expression stability. These algorithms can allow the selection of reference genes that better reflect the intrinsic variability of miRNAs under different health and disease conditions18,43,44 Another strategy involves using non-coding reference genes or synthetic RNAs added as exogenous controls. While these can normalize technical variation in RNA isolation, they do not correct for sample collection fluctuations and cannot improve test accuracy31. Therefore, endogenous genes are often preferred, and algorithms such as GeNorm, NormFinder, and BestKeeper are employed to select the most stable reference genes, increasing the robustness of results16.
In this study, we assessed the stability of each candidate gene and confirmed that snRNA U6 is a suitable reference gene for qRT-PCR analysis in plasma samples from COVID-19 patients. This conclusion was supported by the consistent stability rankings produced by the four statistical algorithms—NormFinder, ΔCt, BestKeeper, and GeNorm—which collectively identified snRNA U6 as the most stable reference gene for this sample set20,45.
The selection of snRNA U6 was particularly relevant, as its stability has been widely reported in various biological due to its more consistent expression across different samples and condition8,25,35,46,47,48,49, supporting its use as a robust internal control in studies of circulating small RNAs.
The use of these algorithms increased the reliability of the analysis, offering a comprehensive assessment of the stability of gene expression and allowing the selection of reference genes that better captured the intrinsic variability of miRNAs in different health conditions. Furthermore, although snRNA U6 is widely described as a reference normalizer in various pathologies10,46,47, there is a scarcity of reports in the literature on the most suitable normalizers for analysis in plasma from COVID-19 patients. So, this study presents snRNA U6 as a stable reference gene for the future differential expression analysis of miRNAs in plasma from COVID-19 patients.
This study provides a novel contribution by demonstrating, for the first time, that snRNA U6 can be utilized as an endogenous reference to normalize gene expression data for circulating miRNAs obtained from plasma of COVID-19 patients. The use of multiple established algorithms ensured a rigorous analysis, underscoring the methodological strength of our approach. However, sample size is a limitation of our study, emphasizing the need for future investigations with larger and more diverse cohorts to confirm the applicability of snRNA U6 across different clinical settings.
Conclusion
In conclusion, this study provides strong evidence for the use of snRNA U6 as a stable reference gene for miRNA expression analysis in plasma samples from COVID-19 patients, addressing the need for reliable internal controls in this setting. The combined use of NormFinder, BestKeeper, ΔCt, and GeNorm algorithms reinforces the robustness of snRNA U6 as a reference gene, enhancing the confidence in its stability across various conditions. Future studies should incorporate larger and more diverse cohorts to further validate the stability of snRNA U6 and investigate the roles of miR-320a and other candidate miRNAs in COVID-19. These findings establish a valuable foundation for future research, potentially improving the accuracy of miRNA biomarker discovery in infectious diseases and contributing to advancements in diagnostic and therapeutic strategies.
Materials and methods
Study design and ethical considerations
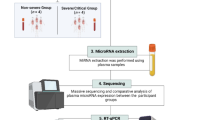
This study was carried out from August 2021 to August 2022 with individuals who tested to SARS-CoV-2 by RT-qPCR at Laboratório de Farmacogenômica e Epidemiologia Molecular (LAFEM), located at Universidade Estadual de Santa Cruz (UESC). LAFEM/UESC worked in partnership with the Central Public Health Laboratory Professor Gonçalo Moniz (LACEN–BA), supporting the routine diagnostics for SARS-CoV-2 detection in the southern region of Bahia State (Fig. 4), which was one of the main epicenters of the COVID-19 pandemic after the capital Salvador and its metropolitan region. After collecting a nasopharyngeal swab for viral detection, peripheral venous blood was also collected from individuals that agree to participate in the study as volunteers. Informed written consent was obtained from all study participants. This study was conducted in accordance with the relevant guidelines and regulations and was approved by the research ethics committee of the State University of Santa Cruz (Universidade Estadual de Santa Cruz), Bahia, Brazil (approval number: CAAE 38627420.3.0000.5526).

Map of Brazil and the state of Bahia, highlighting the area (cities in the southern micro-region) attended by LAFEM/UESC as campaign laboratory to SARS-CoV-2 detection.
Ethical considerations
The study was conducted in accordance with the guidelines of the Declaration of Helsinki and approved by the Research Ethics Committee of the State University of Santa Cruz under registration number CAAE: 38627420.3.0000.5526. Informed consent was obtained from all participants, and all information was treated confidentially and anonymised to guarantee the privacy of the participants.
Groups definition
Individuals with SARS-CoV-2 RNA detectable by RT-qPCR and respiratory symptoms were included in the “case” group. Individuals with undetectable results for SARS-CoV-2 by RT-qPCR and without respiratory symptoms were included in the “control” group. In both groups, only individuals who consented to undergo SARS-CoV-2 testing and venipuncture for blood sample collection were included (Fig. 5).

Flow diagram of the development research processes.
Laboratory detection of SARS-CoV-2 infection
Nasopharyngeal swab specimens were collected from all individuals and subsequently tested at LAFEM/UESC. Viral RNA was extracted using an automated Loccus EXTRACTA 32 device and the MVXA-P016 kit. The Allplex™ 2019-nCoV assay (Seegene®, Seoul, Korea) and the SARS-CoV-2 EDx assay (Bio-Manguinhos, FIOCRUZ, Brazil) were employed to detect SARS-CoV-2 RNA by RT-qPCR, in accordance with the manufacturer’s instructions, utilising the 7500 Fast Real-Time PCR System (Applied BiosystemsTM, Life Technologies, USA). The results were classified as positive (detection of SARS-CoV-2), negative (absent of detection), or inconclusive. Inconclusive results were excluded from the subsequent analysis.
Sample collection for MiRNA expression analysis
Venous blood samples (4 mL) were collected using a 21-gauge needle (Becton-Dickinson, Franklin Lakes, NJ, USA) into tubes containing K2-EDTA. The plasma fraction was separated by centrifugation and 200 µL of the plasma sample was mixed with 600 µL of Trizol LS (Invitrogen) and subsequently stored at − 80 °C for a maximum of 24 h after blood collection.
RNA isolation and quantification
RNA isolation involved adding 200 µL of cold chloroform (~ 8 °C), followed by inversion, room temperature incubation (5 min), and centrifugation (4 °C, 14,000 rpm, 20 min) to separate phases (Fig. 6). The aqueous phase, with an average volume of 600 µL, was collected and stored in a separate collection tube. Subsequently, 1000 µL of absolute isopropanol was added and the mixture was stored at − 20 °C for a period of approximately 12 h. Subsequently, the samples were thawed and subjected to centrifugation at 4 °C and 14,000 rpm for 20 min. The supernatant was discarded, and the pellet was retained for further analysis. Subsequently, 1000 µL of 70% ethanol was added to the tube, after which the sample was subjected to centrifugation at 4 °C and 14,000 rpm for 20 min. Once more, the supernatant was discarded, and the pellet was retained. The tubes containing the pellets were then left to dry at room temperature for approximately 15 min. Subsequently, the pellet was resuspended in 20 µL of RNase-free water and stored at − 80 °C. All reagents were employed at a temperature of approximately 8 °C.
RNA concentration was measured using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Only RNA samples with a 260/280 ratio of at least 1.8 were included in the subsequent analysis. A total of 500 ng of RNA was used for reverse transcription, employing specific primers for microRNAs and a TaqMan microRNA reverse transcription kit (Applied Biosystems) in a reduced volume of 20 µL of reverse transcription reaction, in accordance with the manufacturer’s instructions.
The cDNA synthesis used the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems) with stem-loop primers for snRNU6B, snRNA U6, miR-328-3p, miR-342-3p, and miR-320a (Thermo Fisher Scientific assay IDs: 001093, 001973, 000543, 002260, 002277). Additionally, the following microRNAs were assessed: 42-3p and miR-320a (Thermo Fisher Scientific assay IDs: 001093, 001973, 000543, 002260, and 002277). The reaction was conducted with 5 µL of purified microRNA in a total volume of 20 µL, following the manufacturer’s recommended protocol, which entailed incubation at 16 °C for 30 min, 42 °C for 30 min, and at 85 °C for 5 min.
Each qPCR reaction included 4.5 µL cDNA, 0.5 µL TaqMan 20X Assay, and 10 µL universal PCR master mix (Applied Biosystems) in a 20 µL final volume. Each sample was processed duplicate, with each cycle comprising 95 °C for 3 min, 95 °C for 15 s, and 60 °C for 60 s, using a QuantStudio 3.0 system.

Procedures and main steps of miRNA expression analyses by RT-qPCR.
Selection of candidate reference genes and stability analysis
In the initial phase of the study, we evaluated the expression of five candidate reference genes (snRNU6B, snRNA U6, miR-328-3p, miR-342-3p, and miR-320a) in plasma samples from 24 individuals in the case group (detectable for SARS-CoV-2 infection) and 24 individuals in the control group (non-detectable for SARS-CoV-2 infection). The selection of these candidates was based on the team’s prior experience with the method41,50, as well as a literature review supporting the use of these molecules as endogenous reference genes in miRNA expression studies19,51.
Among them, snRNU6B and snRNA U6 belong to distinct classes of non-coding RNAs with different biological roles: snRNU6B is a small nucleolar RNA involved in ribosomal RNA processing, while snRNA U6 is a small nuclear RNA (snRNA) essential for pre-mRNA splicing as part of the spliceosome complex. The expression levels of these candidates were assessed using one-way ANOVA (p > 0.001) to determine potential differences between the groups.
Consequently, the remaining candidate, which exhibited minimal variation in expression levels between case and control groups (less than 1-fold change), was selected as a reference gene for COVID-19 plasma sample analysis. This careful selection provided a robust basis for subsequent analyses. Stability and consistency of miRNA expression were evaluated using specialized software, including RefFinder52 and NormFinder53, to ensure reliable results. NormFinder, an Excel-based tool, applies a model-based approach that assigns stability values to candidate normalizers based on intra- and inter-group variation, with lower values indicating greater stability. RefFinder, an online application, integrates four commonly used algorithms—Bestkeeper54, comparative ΔCt55, NormFinder53 and GeNorm—offering a comprehensive assessment to identify the most stable gene or gene pair for normalization. All statistical analyses were conducted with GraphPad Prism 5.0, considering p < 0.05 as statistically significant (t-test).
Data availability
The datasets generated during and/or analyzed during the current study are not publicly but are available from the corresponding author on reasonable request.
References
Cevik, M., Kuppalli, K., Kindrachuk, J. & Peiris, M. Virology, transmission, and pathogenesis of SARS-CoV-2. BMJ 371, 1–6 (2020).
Eastin, C. & Eastin, T. Clinical characteristics of coronavirus disease 2019 in China. J. Emerg. Med. 58, 711–712 (2020).
Araf, Y. et al. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 94, 1825–1832 (2022).
Mistry, P. et al. SARS-CoV-2 variants, vaccines, and host immunity. Front. Immunol. 12, 809244 (2022).
Santos, C. V. B. et al. The effectiveness of COVID-19 vaccines against severe cases and deaths in Brazil from 2021 to 2022: A registry-based study. Lancet Reg. Health – Am. 20, 1–13 (2023).
Kaplan, G. G. et al. The multiple waves of COVID-19 in patients with inflammatory bowel disease: A temporal trend analysis. Inflamm. Bowel Dis. 28, 1687–1695 (2022).
Panda, M. et al. MiRNA-SARS-CoV-2 dialogue and prospective anti-COVID-19 therapies. Life Sci. 305, 1–21 (2022).
Yao, Q., Chen, Y. & Zhou, X. The roles of MicroRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 51, 11–17 (2019).
Yu, X., Odenthal, M. & Fries, J. W. U. Exosomes as MiRNA carriers: Formation–function–future. Int. J. Mol. Sci. 17, 1–12 (2016).
Zhou, G. et al. Identification of miR-199a-5p in serum as noninvasive biomarkers for detecting and monitoring osteosarcoma. Tumour Biol. J. Int. Soc. Oncodevelopment. Biol. Med. 36, 8845–8852 (2015).
Weber, J. A. et al. The MicroRNA spectrum in 12 body fluids. Clin. Chem. 56, 1733–1741 (2010).
McDermott, A. M., Kerin, M. J. & Miller, N. Identification and validation of MiRNAs as endogenous controls for RQ-PCR in blood specimens for breast cancer studies. PloS One. 8, e83718 (2013).
Niu, Y. et al. Identification of reference genes for Circulating MicroRNA analysis in colorectal cancer. Sci. Rep. 6, 35611 (2016).
Chien, H. Y. et al. Circulating MicroRNA as a diagnostic marker in populations with type 2 diabetes mellitus and diabetic complications. J. Chin. Med. Assoc. JCMA. 78, 204–211 (2015).
Satake, E. et al. Circulating MiRNA profiles associated with hyperglycemia in patients with type 1 diabetes. Diabetes 67, 1013–1023 (2018).
Vavassori, C., Cipriani, E. & Colombo, G. I. Circulating MicroRNAs as novel biomarkers in risk assessment and prognosis of coronary artery disease. Eur. Cardiol. Rev. 17, 1–11 (2022).
Liang, Y. et al. Circulating MicroRNAs as emerging regulators of COVID-19. Theranostics 13, 125–147 (2023).
Faraldi, M. et al. Normalization strategies differently affect circulating MiRNA profile associated with the training status. Sci. Rep. 9, 1–13 (2019).
Donati, S., Ciuffi, S. & Brandi, M. L. Human circulating MiRNAs Real-time qRT-PCR-based analysis: An overview of endogenous reference genes used for data normalization. Int. J. Mol. Sci. 20, 4353 (2019).
Xie, F., Wang, J. & Zhang, B. RefFinder: A web-based tool for comprehensively analyzing and identifying reference genes. Funct. Integr. Genom. 23, 125 (2023).
de Torso, N. Data normalization of urine MiRNA profiling from head and neck Cancer patients treated with cisplatin. Int. J. Mol. Sci. 24, 1–15 (2023).
Zhang, Y., Tang, W., Peng, L., Tang, J. & Yuan, Z. Identification and validation of MicroRNAs as endogenous controls for quantitative polymerase chain reaction in plasma for stable coronary artery disease. Cardiol. J. 23, 694–703 (2016).
Franco, S., Llibre, J. M., Jou, T., Tural, C. & Martínez, M. A. Normalization of circulating plasma levels of MiRNAs in HIV-1/HCV co-infected patients following direct-acting antiviral-induced sustained virologic response. Heliyon 9, 1–10 (2023).
Giannella, A. et al. Circulating MicroRNA signatures associated with disease severity and outcome in COVID-19 patients. Front. Immunol. 13, 1–21 (2022).
Se, B. et al. Identification of MiR-93 as a suitable MiR for normalizing MiRNA in plasma of tuberculosis patients. J. Cell. Mol. Med. 19, 1606–1613 (2015).
W, K. et al. Identification of circulating miR-22-3p and miR-93-5p as stable endogenous control in tuberculosis study. Diagn. Basel Switz. 10, 868–881 (2020).
Zhu, H. T. et al. Identification of suitable reference genes for qRT-PCR analysis of circulating MicroRNAs in hepatitis B virus-infected patients. Mol. Biotechnol. 50, 49–56 (2012).
Tang, G. et al. Different normalization strategies might cause inconsistent variation in circulating MicroRNAs in patients with hepatocellular carcinoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 21, 617 (2015).
Klimczak, D. et al. Plasma microRNA-155-5p is increased among patients with chronic kidney disease and nocturnal hypertension. J. Am. Soc. Hypertens. JASH. 11, 831–841e4 (2017).
Gevaert, A. B. et al. MicroRNA profiling in plasma samples using qPCR arrays: Recommendations for correct analysis and interpretation. PLoS ONE 13, 1–13 (2018).
Lamba, V., Ghodke-Puranik, Y., Guan, W. & Lamba, J. K. Identification of suitable reference genes for hepatic MicroRNA quantitation. BMC Res. Notes 7, 129 (2014).
Martens-Uzunova, E. S., Olvedy, M. & Jenster, G. Beyond microRNA–novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett. 340, 201–211 (2013).
Kaija, H., Pakanen, L. & Porvari, K. RNU6B, a frequent reference in MiRNA expression studies, differentiates between deaths caused by hypothermia and chronic cardiac ischemia. Int. J. Legal Med. 134, 159–162 (2020).
Didychuk, A. L., Butcher, S. E. & Brow, D. A. The life of U6 small nuclear RNA, from cradle to grave. RNA 24, 437–460 (2018).
Duan, Z. Y. et al. U6 can be used as a housekeeping gene for urinary sediment MiRNA studies of IgA nephropathy. Sci. Rep. 8, 10875 (2018).
Fiedler, S. D., Carletti, M. Z. & Christenson, L. K. Quantitative RT-PCR methods for mature MicroRNA expression analysis. Methods Mol. Biol. Clifton NJ. 630, 49–64 (2010).
Hao, X., Xin, R. & Dong, W. Decreased serum Exosomal miR-320a expression is an unfavorable prognostic factor in patients with hepatocellular carcinoma. J. Int. Med. Res. 48, 1–11 (2020).
Villard, A., Marchand, L., Thivolet, C. & Rome, S. Diagnostic value of cell-free circulating MicroRNAs for obesity and type 2 diabetes: A meta-analysis. J. Mol. Biomark. Diagn. 6, 251 (2015).
Zhong, Y., Zhu, F. & Ding, Y. Differential MicroRNA expression profile in the plasma of preeclampsia and normal pregnancies. Exp. Ther. Med. 18, 826 (2019).
Sommariva, E. et al. MiR-320a as a potential novel Circulating biomarker of arrhythmogenic cardiomyopathy. Sci. Rep. 7, 1–10 (2017).
Prado, M. S. G., de Jesus, M. L., de Goes, T. C., Mendonça, L. S. O. & Kaneto, C. M. Downregulation of Circulating miR-320a and target gene prediction in patients with diabetic retinopathy. BMC Res. Notes. 13, 155 (2020).
Mestdagh, P. et al. A novel and universal method for MicroRNA RT-qPCR data normalization. Genome Biol. 10, 1–10 (2009).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, 1–12 (2002).
Dong, J., Li, J., Huang, J. & Wu, J. Identification of suitable reference genes for MiRNA quantitation in bumblebee (Hymenoptera: Apidae) response to reproduction. Apidologie 50, 40–50 (2019).
Causin, R. L. et al. Identification and performance evaluation of housekeeping genes for MicroRNA expression normalization by reverse transcription-quantitative PCR using liquid-based cervical cytology samples. Oncol. Lett. 18, 4753 (2019).
Papadaki, C. et al. Circulating MicroRNAs regulating DNA damage response and responsiveness to cisplatin in the prognosis of patients with non-small cell lung cancer treated with first-line platinum chemotherapy. Cancers 12, 1282 (2020).
Wang, L. et al. Differential expression of plasma miR-146a in sepsis patients compared with non-sepsis-SIRS patients. Exp. Ther. Med. 5, 1101–1104 (2013).
Madadi, S. & Soleimani, M. U6 as a MicroRNA normalizer in serum of patients with hepatocellular carcinoma. Ann. Clin. Biochem. 56, 623–623 (2019).
Zhang, Y. et al. Plasma MicroRNA-122 as a biomarker for viral-, alcohol-, and chemical-related hepatic diseases. Clin. Chem. 56, 1830–1838 (2010).
Prado, M. S. G. et al. Identification of miR-328-3p as an endogenous reference gene for the normalization of MiRNA expression data from patients with diabetic retinopathy. Sci. Rep. 9, 19677 (2019).
Nascimento, N. P. G., Gally, T. B., Borges, G. F., Campos, L. C. G. & Kaneto, C. M. Systematic review of circulating MICRORNAS as biomarkers of cervical carcinogenesis. BMC Cancer. 22, 862 (2022).
Xie, F., Xiao, P., Chen, D., Xu, L. & Zhang, B. MiRDeepFinder: A MiRNA analysis tool for deep sequencing of plant small RNAs. Plant. Mol. Biol. 80, 75–84 (2012).
Andersen, C. L., Jensen, J. L. & Ørntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and Colon cancer data sets. Cancer Res. 64, 5245–5250 (2004).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515 (2004).
Silver, N., Best, S., Jiang, J. & Thein, S. L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 7, 33 (2006).
Acknowledgements
We thank the Programa de Pós-graduação em Biologia e Biotecnologia de Microrganismos (PPGBBM) and the Programa de Pós-graduação em Ciências da Saúde (PPGCS).
Funding
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES); Programa Laboratórios de Campanha do Ministério de Ciência, Tecnologia e Inovação - MCTI/FINEP (0494/20 − 01.20.0026.00); Fundação de Amparo à Pesquisa do Estado da Bahia - FAPESB (Edital 02/2020 - Programa Pesquisa para o SUS: gestão compartilhada em saúde - PPSUS) and Universidade Estadual de Santa Cruz (UESC).
Author information
Authors and Affiliations
Contributions
All authors contributed to the development and completion of this study. Silva, ITSS, conceived and designed the research, Fehlberg HF, Ferreira FB, Silva MF, Santos PR, Vinicius MP, performed the experiments and data collection. Silva ITSS, Dias JCT Albuquerque GA, Mariano APM, Gadelha SR and Kaneto CM, performed data analysis and interpretation. Silva ITSS, Gadelha SR and Kaneto CM, contributed to the writing and editing of the manuscript. All authors reviewed and approved the final version of the manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
de Santana Silva, Í.T.S., Fehlberg, H.F., Ferreira, F.B. et al. Identification of SnRNA U6 as an endogenous reference gene for normalization of MiRNA expression data in COVID-19 patients. Sci Rep 15, 26636 (2025). https://doi.org/10.1038/s41598-025-04062-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-04062-9
