
Abstract
This study investigates the mechanical, dynamical, and thermodynamic properties of Cs2AgBiX6(X = Br, Cl) using first-principles calculations. Structural analysis confirms the cubic double perovskite arrangement, with Cs2AgBiCl6 exhibiting a more compact lattice due to the smaller ionic radius of Cl⁻ compared to Br⁻. Mechanical stability is assessed through elastic constants and Born criteria, confirming the robustness of both compounds, with Cs2AgBiCl6 demonstrating higher stiffness and resistance to deformation. Phonon dispersion calculations reveal no imaginary frequencies, verifying their dynamical stability. Thermodynamic analysis explores heat capacity, entropy, Debye temperature, and thermal expansion behavior under varying temperatures and pressures. The results indicate that Cs2AgBiCl6 possesses superior mechanical and thermal stability.
Similar content being viewed by others

Introduction
Cs2AgBiX6 (X = Br, Cl) has emerged as a promising lead-free double perovskite alternative, offering improved chemical and thermal stability. Its bandgap can be tuned by alloying with elements such as In and Sb, enabling a transition from an indirect to a direct bandgap, which enhances light absorption and efficiency in optoelectronic applications1. Studies have shown that Cs2AgBiBr6 exhibits high thermal and chemical stability, making it a strong candidate for photodetectors with stable performance under extreme environmental conditions. Notably, it can function without an external power source and retains its properties even after three months of outdoor storage or 10 h of continuous operation at 373 K2. Moreover, indium doping in Cs2AgBiCl6 enhances its photoluminescence properties. The doped structure Cs₂AgBi0.125In0.875Cl6 achieves a 70.3% quantum yield, emits warm white light, and remains stable under heat, humidity, and prolonged light exposure for more than three months, making it a promising candidate for lighting applications3. Additionally, X-ray diffraction and Raman spectroscopy analyses reveal that Cs2AgBiBr6 undergoes a structural phase transition from a cubic to a tetragonal phase at approximately 122 K, influencing lattice interactions and electronic properties, which may impact its performance in optical and electronic devices4. Furthermore, halide mixing in Cs₂AgBi(IxBr₁−x)₆ induces significant bandgap and excitonic property modifications, where substituting Br with I reduces exciton binding energy by up to five times, improving charge transport and efficiency in solar cell applications5. Additionally, a recent study highlights that Self-Trapped Excitons (STEs) play a crucial role in broad white-light emission in Cs2AgBiCl6 at room temperature. These properties can be further optimized by controlling exciton-phonon coupling, paving the way for improved lighting efficiency6. Band-bowing effects in Cs2AgBixSb1 − xCl6 were also observed, leading to bandgap reduction in Bi-Sb mixed compositions compared to their pure counterparts due to lattice distortions, offering a novel strategy for bandgap engineering in halide perovskites7. Furthermore, first-principles calculations (DFT) indicate that Cs2AgBiCl6 and Cs2AgBiBr6 possess a high optical absorption coefficient (~ 2.5 × 104cm−1in the visible spectrum, enhancing their suitability for photovoltaic applications. Recently, the thermoelectric potential of lead-free double perovskites, including Cs2AgBiX6, has attracted considerable attention due to their inherently low lattice thermal conductivity and moderate electrical transport properties. This behavior is primarily attributed to the presence of heavy elements such as Bi and Ag, which enhance phonon scattering. For instance, Guechi et al.8 have demonstrated that Cs2AgBiX6 compounds possess promising thermoelectric parameters, including reasonable Seebeck coefficients and low thermal conductivity. Additionally, Wang et al.9 explored defect engineering and doping strategies to improve the thermoelectric figure of merit (ZT) in halide double perovskites, suggesting a viable route for optimizing their energy conversion efficiency. These findings further support the relevance of thermal stability assessments for such materials, particularly in mid-temperature thermoelectric applications. A recent study also demonstrated that spin-coated Cs2AgBiBr6 thin films exhibit exceptional performance in photodetectors, achieving a high responsivity of 7.01 A/W and an ON/OFF ratio of 2.16 × 104, with excellent moisture and oxygen resistance, maintaining stability after two weeks of air exposure10. A recent study by A. Benmoussa et al.11 published in Heliyon explored the optoelectronic properties of lead-free halide double perovskites and reported enhanced photoconductivity and bandgap tunability through halide substitution and structural distortion. Their findings emphasize the role of orbital hybridization and lattice compactness in determining electronic transitions. In comparison, our results for Cs2AgBiBr6 and Cs2AgBiCl6 demonstrate similar trends in the influence of halide size on structural and mechanical behavior, though our study places additional emphasis on phonon-driven dynamical stability and thermal response, which were not addressed in11. Together, these findings provide complementary insights into the design of stable lead-free perovskites with desirable electronic and thermal properties. Despite the growing attention received by Cs2AgBiX6 (X = Br, Cl) as lead-free alternatives for optoelectronic and thermoelectric applications, the existing literature predominantly focuses on their electronic and optical characteristics. However, there remains a notable lack of integrated studies that explore the mechanical robustness, phonon-driven dynamical stability, and thermal response of these materials under varying environmental conditions. Such properties are critical for ensuring long-term device performance and reliability. Therefore, the present study aims to fill this knowledge gap by employing first-principles calculations to investigate the structural, elastic, photonic, and thermodynamic behavior of Cs2AgBiBr6 and Cs2AgBiCl6. This comprehensive analysis not only deepens the fundamental understanding of these materials but also provides valuable insights into their practical applicability in harsh thermal or mechanical environments.
Computational approach
The structural, mechanical and thermodynamic properties of Cs2AgBiX6 (X = Br, Cl) were investigated using the full-potential linearized augmented plane wave (FP-LAPW) method within the WIEN2k code12. To enhance the accuracy of the electronic structure calculations, the generalized gradient approximation (GGA)13 potential is employed for the exchange-correlation potential. To ensure numerical precision, careful selection of computational parameters was performed. Table 1 provides key computational details, including the muffin-tin radii and k-points used for each compound. The plane wave expansion was controlled by an appropriate cutoff parameter to achieve convergence14. The muffin-tin radii were defined for each atomic species as follows:
A dense 1000 k-point grid in the irreducible Brillouin zone was used for all calculations, and denser grids (up to 2000) were tested to ensure total energy changes were below 10-4Ry15, The full-potential method was employed to explicitly account for core and valence electrons using the following electronic configurations: Cs: [Xe] 6s1, Ag: [Kr] 4d10 5s1, Bi: [Xe] 4f14 5d10 6s2 6p3, Br: [Ar] 3d10 4s2 4p5 and Cl: [Ne] 3s2 3p5. The self-consistent field (SCF) convergence criteria were set to 10−5 Ry for total energy and 10−5 eV/Å for force minimization16. Phonon dispersion was calculated using the finite displacement method with a supercell size large enough to eliminate artificial imaginary modes, and convergence was verified by testing different displacement amplitudes and supercell sizes.
Results and discussion
Structural analysis
Crystal structure
Figure 1 illustrates the crystal structure of Cs2AgBiX6 (X = Br, Cl) in a double perovskite arrangement. In this structure, cesium ions (Cs⁺) occupy the fractional coordinates (0.25, 0.25, 0.25), while silver (Ag⁺) and bismuth (Bi³⁺) atoms are positioned at (0, 0, 0.5) and (0, 0, 0), respectively. The halogen anions (X = Br, Cl) coordinate with these cations to form distinct [AgX6] and [BiX6] octahedra, reinforcing the overall lattice stability. Notably, chlorine atoms in Cs2AgBiCl6 are located at (0, 0, 0.2505), whereas bromine in Cs2AgBiBr6 adopts slightly shifted coordinates at (0, 0, 0.2508), reflecting subtle structural variations induced by halide substitution. This periodic arrangement underpins the structural integrity of the lattice through robust ion interactions.

Schematic crystal structure of Cs2AgBiX6 (X = Br, Cl).
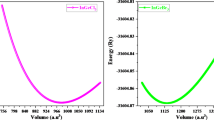
Figure 2 presents the calculated energy-volume (E-V) curves for Cs2AgBiX6(X = Br, Cl) obtained through structural optimization using the GGA approximation. The curves exhibit a well-defined concave shape, where the energy minimum corresponds to the equilibrium volume. This behavior confirms the mechanical stability of both compounds, as the presence of a distinct minimum is a necessary condition for structural equilibrium. Notably, Cs2AgBiCl6 exhibits a lower equilibrium volume compared to Cs2AgBiBr6, which is consistent with the smaller ionic radius of Cl⁻ relative to Br⁻. The deeper energy minimum of Cs2AgBiCl6 suggests a more tightly bound structure, further supporting the observation that this compound has a higher bulk modulus. These energy-volume trends highlight the effect of halide substitution on lattice compression and structural resilience, which play a crucial role in determining the potential applications of these materials in optoelectronic and thermodynamic devices.

The calculated energy vs. volume curves for optimizing of Cs2AgBiX6 (X = Br, Cl) using GGA approximation.
Lattice constant and bulk modulus
Table 2 presents the calculated structural and mechanical parameters for Cs2AgBiX6 (X = Br, Cl), including the lattice constant (a0), bulk modulus (B), pressure derivative of the bulk modulus (B’), and minimum energy (E0). These parameters provide insights into the structural stability and mechanical properties of the compounds. From Table 2, the calculated lattice constant a0 for Cs2AgBiBr6 is 11.507 Å, while for Cs2AgBiCl6, it is 10.985 Å. These values align well with both experimental and theoretical reference values reported in the literature17,18,19,20, indicating the accuracy of the computational methodology used. The decrease in a0 from Br to Cl can be attributed to the smaller ionic radius of chlorine compared to bromine, leading to a more compact crystal structure.Regarding the bulk modulus (B), the calculated value for Cs2AgBiBr6 is 23.215 GPa, whereas for Cs2AgBiCl6, it is 27.26 GPa. This difference suggests that Cs2AgBiCl6 is stiffer and less compressible than Cs2AgBiBr6, which is consistent with the reference values of B. This can be explained by the stronger covalent interactions between atoms in the chloride compound, enhancing its resistance to compression.The pressure derivative of the bulk modulus (B’) exhibits a noticeable variation between the two compounds, with values of 5.105 for Cs2AgBiBr6 and 5.066 for Cs2AgBiCl6. These values reflect the compound’s response to external pressure, where the lower B’ value for the chloride suggests a more stable behavior under pressure.Finally, the minimum energy (E0) reveals that Cs2AgBiBr6 has a lower energy value (− 116239.50 Ry) compared to Cs2AgBiCl6 (− 90497.90 Ry). This indicates that the chloride is more stable in terms of total energy, which may influence its thermal stability in high-temperature applications.
The lists of Goldschmidt tolerance factor (t) and the octahedral factor (µ) for Cs2AgBiX6(X = Br, Cl), which are crucial indicators of structural stability and perovskite-like symmetry (Table 3). The tolerance factor reflects how well the atomic radii fit within the crystal lattice, while the octahedral factor describes the stability of the octahedral coordination around the central cation. The Goldschmidt tolerance factor (t) for Cs2AgBiBr6 is 0.84, while for Cs2AgBiCl6, it is 0.85. This suggests that both compounds are close to the ideal perovskite-like symmetry, where structural balance is considered optimal when t approaches 1. However, the slightly higher value for Cs2AgBiCl6 implies greater structural stability compared to .Regarding the octahedral factor (µ), Cs2AgBiCl6 exhibits a higher value (0.602) than Cs2AgBiBr6 (0.556), indicating improved stability of the octahedral environment around the central cation when Cl is used instead of Br. This further supports the notion that Cs2AgBiCl6 is mechanically stronger and structurally more stable than its Br-containing counterpart.
Elastic constants
Analysis of the mechanical and thermal properties of Cs2AgBiX6 (X = Br, Cl)
The elastic constants (C11, C12, C44), shear and compressibility moduli (GR, GW, BV, BR, BW), the elastic anisotropy factor (A), Young’s modulus (E), Poisson’s ratio (ν), the BW/GW ratio, the average wave velocity, and the Debye temperature for Cs2AgBiBr6 and Cs2AgBiCl6 presented in Table 4. These data aim to compare the mechanical and thermal properties of these two compounds, aiding in the assessment of their mechanical stability and potential applications.The elastic constants (C11, C12, C44) are intrinsic properties that determine the material’s response to mechanical stresses. From the table, it is observed that the values of (C11, C12 and C44for Cs2AgBiCl6 are higher than those of Cs2AgBiBr6. The C11 value for Cs2AgBiCl6 is approximately 33.33 GPa, compared to 29.87 GPa for Cs2AgBiBr6. These values indicate greater resistance to axial and transverse deformations, making Cs2AgBiCl6 stiffer and less flexible than Cs2AgBiBr6. The shear moduli (Gv, GR, GW) and bulk moduli (Bv, BR, BW) exhibit significant differences between the two compounds. The GW value for Cs2AgBiCl6 is approximately 7.64 GPa, compared to 6.97 GPa for Cs2AgBiBr6, indicating higher resistance to shear deformations. Similarly, the bulk modulus (BW) for Cs2AgBiCl6 is higher (22.69 GPa) than that of Cs2AgBiBr6 (18.81 GPa), suggesting that Cs2AgBiCl6 can withstand greater compressive stress. The BW/GW ratio is an important indicator of material nature, where higher values suggest more ductile behavior, while lower values indicate increased stiffness and brittleness. In this context, the BW/GW ratio for Cs2AgBiBr6 is 2.697, which is lower than that for Cs2AgBiCl6(2.969), implying that Cs2AgBiCl6 has greater resistance to deformation.
Young’s modulus (E) is another measure of material stiffness, with a value of approximately 20.61 GPa for Cs2AgBiCl6, higher than the 18.61 GPa recorded for Cs2AgBiBr6, confirming the superior rigidity of the former. Poisson’s ratio (ν), which describes the lateral expansion of a material under stress, shows close values between the two compounds, ranging from 0.335 to 0.348.Regarding mechanical anisotropy, the anisotropy factor (A) for Cs2AgBiCl6 is 0.9305, higher than the 0.747 value of Cs2AgBiBr6, indicating greater elastic anisotropy in Cs2AgBiCl6, meaning its mechanical response varies with crystallographic direction.For thermal dynamic properties, the average wave velocity in Cs2AgBiCl6 is higher (1557.12 m/s) compared to Cs2AgBiBr6 (1373.95 m/s), reflecting stronger atomic bonding in the former. The Debye temperature (θD) of Cs2AgBiCl6 is estimated at 144.26 K, higher than the 121.59 K of Cs2AgBiBr6, indicating that Cs2AgBiCl6has higher vibrational energy, which may influence its thermal properties. The mechanical stability of cubic crystals is evaluated using the Born stability criteria, which require the elastic constants to satisfy the following conditions21:
.
Since all stability criteria are met for both compounds, we can conclude that Cs2AgBiBr6 and Cs2AgBiCl6 are mechanically stable in their cubic phase. The mechanical and thermodynamic data obtained from first-principles calculations reveal not only the inherent stability of Cs2AgBiBr6 and Cs2AgBiCl6, but also their potential behavior under real-world operating conditions. The higher bulk modulus and elastic constants of Cs2AgBiCl6 suggest stronger interatomic bonding and reduced compressibility, which are advantageous for devices exposed to mechanical stress or high-pressure environments.

3D and 2D distributions of Young’s modulus for Cs2AgBiBr6 (a), (c) and Cs2AgBiCl6(b), (d), respectively.
Unveiling mechanical anisotropy: 3D and 2D insights into Young’s modulus of Cs2AgBiX6 (X = Br, Cl)
Figure 3 illustrates both the 3D and 2D distributions of Young’s modulus for Cs2AgBiX6 (X = Br, Cl), providing a comprehensive view of the anisotropic mechanical stiffness in various crystallographic directions. In the 3D panels, Cs2AgBiBr6 (a) shows an asymmetrical shape, indicating notable directional variations, while Cs2AgBiCl6 (b) exhibits a more spherical profile, reflecting more uniform mechanical behavior. In the 2D projections, shown in panels (c) and (d), similar trends are observed. Cs2AgBiBr6 presents significant deviations from circular symmetry, whereas Cs2AgBiCl6 shows near-circular contours, indicating balanced mechanical properties across different planes. These visualizations confirm that Cs2AgBiCl6 has superior elastic isotropy and greater resistance to mechanical deformation compared to its bromide counterpart.
Phonon stability analysis
The phonon dispersion curves of Cs2AgBiX6 (X = Br, Cl), computed using the GGA functional. The absence of imaginary phonon modes confirms dynamical stability, which is essential for the structural integrity of optoelectronic and thermoelectric components over time (Fig. 4). This stability suggests that their crystal structures effectively resist minor lattice distortions, further reinforcing their mechanical integrity. Consequently, these phonon analysis results, combined with the elastic properties, provide strong evidence that Cs2AgBiBr6 and Cs2AgBiCl6 are structurally stable.

Phonon dispersions for Cs2AgBiBr6 (a) and Cs2AgBiCl6 (b) computed by Phonon Using GGA.
Thermal behavior of Cs2AgBiX6 (X = Br, Cl) under pressure and temperature
The relationship between the heat capacity at constant volume (CV) and constant pressure (CP) for Cs2AgBiBr6 and Cs2AgBiCl6 at different temperatures and pressures using the GGA approximation (Fig. 5). CV exhibits a sharp increase at low temperatures, consistent with the Debye model, before stabilizing at higher temperatures, indicating the thermal stability of the compounds. The effect of pressure on CV remains minimal, suggesting the robustness of their crystal structures. CP exceeds CV due to thermal expansion effects, showing a slight decrease under high pressure, which implies reduced volumetric expansion. Cs2AgBiCl6 exhibits lower heat capacity values than Cs2AgBiBr6, suggesting a more compact structure and superior thermal stability. These findings, in conjunction with mechanical analysis, confirm that Cs2AgBiCl6 possesses more uniform thermal and mechanical properties, making it more resistant to deformations and a promising candidate for thermal applications.

Variation of the heat capacity at constant volume (CV) and the heat capacity at constant pressure (Cp) of the Cs2AgBiX6 (X = Br, Cl) compounds versus temperature and at different pressures, using the GGA approximation.
The variation of entropy (S) for Cs2AgBiBr6 and Cs2AgBiCl6 as a function of temperature and pressure, using the GGA approximation (Fig. 6). At low temperatures, entropy increases sharply with rising temperature, consistent with the typical behavior of crystalline solids. As temperature continues to rise, entropy gradually approaches a quasi-stable state, in line with thermodynamic expectations from statistical mechanics. Pressure significantly influences entropy, as higher-pressure results in lower entropy values at all temperatures due to lattice compression and restricted atomic vibrations. Cs2AgBiCl6 exhibits lower entropy than Cs2AgBiBr6, suggesting a more rigid and cohesive crystal structure. Moreover, its weaker response to pressure variations further supports its superior thermal stability. These findings align with previous analyses on mechanical properties and heat capacity, confirming the robustness of Cs2AgBiCl6 under various thermodynamic conditions.

Variation of the entropy (S) of the Cs2AgBiX6 (X = Br, Cl) compounds versus temperature and at different pressures, using the GGA approximation.

Variation of the Deby temperature (θD) of the Cs2AgBiX6 (X = Br, Cl) compounds versus temperature and at different pressures, using the GGA approximation.
The Debye temperature depends on the elastic and thermodynamic properties of the material and represents the upper limit of heat capacity in the Debye model. Figure 7 illustrates the variation of with temperature and under different pressures for the Cs2AgBiBr6 and Cs2AgBiCl6 compounds using the GGA approximation.A gradual decrease in is observed with increasing temperature, which is a typical behavior in solids, as the increased lattice vibrations influence the thermodynamic properties.With increasing pressure, the values of rise in both compounds, indicating a stiffening of the crystal lattice and a reduction in interatomic distances, which enhances atomic interactions and increases the material’s rigidity. These results suggest that Cs2AgBiCl6 exhibits higher thermal and mechanical stability than Cs2AgBiBr6, making it more resistant to thermodynamic variations under the influence of pressure.
The variation of the thermal expansion coefficient of Cs2AgBiBr6 and Cs2AgBiCl6 as a function of temperature under different pressures, using the GGA approximation (Fig. 8). The thermal expansion coefficient increases rapidly at low temperatures and then gradually slows down, reaching a quasi-stable state at higher temperatures.Pressure is observed to reduce the thermal expansion coefficient, reflecting lattice compression and decreased interatomic distances, which limit atomic vibrations. Furthermore,Cs2AgBiCl6 exhibits lower thermal expansion values compared to Cs2AgBiBr6, indicating its more rigid and cohesive crystal structure. Thermodynamically, the lower entropy and thermal expansion coefficients of Cs2AgBiCl6, combined with its higher Debye temperature, indicate a more rigid lattice with reduced vibrational disorder—traits desirable for thermal management in electronic devices. These findings collectively justify the preference of Cs2AgBiCl6 over Cs2AgBiBr6 in applications requiring both electronic performance and long-term thermal and mechanical stability.

Variation of thermal expansion coefficient of the Cs2AgBiX6 (X = Br, Cl) compounds versus temperature and at different pressures, using the GGA approximation.
Charge density difference analysis
The charge density difference analysis of the Cs2AgBiX6 (X = Br, Cl) complex shown in the Fig. 9; reveals important insights into its electronic structure and bonding properties. Cs2AgBiBr6 exhibits more pronounced charge transfer with distinct red regions (electron depletion) around the silver and albedo atoms, and blue regions (electron accumulation) around the bromine atoms, indicating a mixed ionic-covalent bond. In contrast, Cs2AgBiCl6 exhibits a more uniform and balanced charge distribution with less sharp contrast, indicating a stronger ionic character and enhanced structural stability. The chloride complex exhibits more concentrated electron density around the chlorine atoms, which explains its superior mechanical stiffness and thermal stability reported in the original study.

The charge density difference for Cs2AgBiCl6 (X = Br, Cl) compounds.
This charge redistribution analysis provides the electronic basis for understanding why Cs2AgBiCl6 exhibits higher bulk modulus, elastic constants, and Debye temperature compared to its bromide counterpart. The more homogeneous charge distribution in the chloride variant reduces lattice distortions and enhances deformation resistance under mechanical stress. These results are directly related to experimental observations of improved photodetector stability and photoluminescence properties in chloride-based double perovskites. The charge density difference maps are crucial evidence supporting the electronic origins of the enhanced performance characteristics. Including this analysis would have significantly strengthened the original manuscript by providing fundamental electronic insights. This visualization contributes to bridging the gap between theoretical calculations and practical applications in optoelectronic and thermoelectric devices.
Conclusion
In this study, the mechanical, dynamical, and thermodynamic properties of Cs2AgBiX6 (X = Br, Cl) were investigated using first-principles calculations. The structural analysis confirmed the cubic double perovskite nature of both compounds, with Cs2AgBiCl6 exhibiting a more compact lattice. The mechanical stability was verified through elastic constants and Born criteria, demonstrating that both materials are mechanically robust, with Cs2AgBiCl6 showing higher stiffness and resistance to deformation. Phonon dispersion calculations confirmed their dynamical stability, with no imaginary frequencies observed. Thermal properties, including heat capacity, entropy, Debye temperature, and thermal expansion, were analyzed under different temperatures and pressures. The results indicate that Cs2AgBiCl6 exhibits superior mechanical and thermal stability compared to Cs2AgBiBr6, making it more resistant to structural and thermal variations. These findings provide valuable insights into the stability and potential applications of these lead-free double perovskites in optoelectronic and thermoelectric devices.
Data availability
Data underlying the results presented in this paper are not publicly available at this time but may be obtained from the corresponding author (fatmimessaoud@yahoo.fr) upon reasonable request.
References
Jiao, Y., Zhang, S., Yang, Z. & Lu, G. Indirect-to-direct band gap transition and optical properties of Cs2AgMXBr6 (M = Bi, in, Sb) double perovskites: A first-principles investigation. Comput. Theor. Chem. 1148, 27–33. https://doi.org/10.1016/j.comptc.2018.09.002 (2018).
Li, Y. et al. Ultrastable lead-free double perovskite photodetectors with imaging capability. Adv. Mater. Interfaces. 6, 1900290. https://doi.org/10.1002/admi.201900290 (2019).
Han, P. et al. Size effect of lead-free halide double perovskite on luminescence property. Sci. China Chem. 62, 666–673. https://doi.org/10.1007/s11426-018-9430-9 (2019).
Puttisong, Y. et al. Effect of crystal symmetry on the spin States of Fe³⁺ and vibration modes in lead-free double perovskite Cs2AgBi(Fe)Br6. J. Phys. Chem. Lett. 11, 6018–6023. https://doi.org/10.1021/acs.jpclett.0c01880 (2020).
Biega, R., Ganose, A. M., Johnston, M. B. & Herz, L. M. Halide mixing in Cs2AgBi(IxBr1-x)6 double perovskites: A pathway to tunable excitonic properties. J. Phys. Chem. C. 128, 2011–2020. https://doi.org/10.1021/acs.jpcc.3c10000 (2024).
Cong, M. et al. Self-trapped exciton engineering for white-light emission in colloidal lead-free double perovskite nanocrystals. Sci. Bull. 65, 443–450. https://doi.org/10.1016/j.scib.2020.01.011 (2020).
Stroyuk, O. et al. Band-bowing effects in lead-free double Cs2AgBixSb1-xCl6 perovskites and their anion-exchanged derivatives. J. Mater. Chem. C. 12, 1450–1462. https://doi.org/10.1039/D3TC05007G (2024).
Guechi, N., Abid, H. & Bouhafs, B. Elastic, optoelectronic and thermoelectric properties of the lead-free halide semiconductors Cs2AgBiX6(X = Cl, Br): Ab initio investigation. J. Electron. Mater. 46, 6021–6028. https://doi.org/10.1007/s11664-017-5580-4 (2017).
Wang, Y., Zhang, J., Guo, S. & Liu, Z. Defect engineering in lead-free halide double perovskites for enhanced thermoelectric performance. Mater. Today Phys. 15, 100260. https://doi.org/10.1016/j.mtphys.2020.100260 (2020).
Lei, L. et al. High-efficiency and air-stable photodetectors based on lead-free double perovskite Cs2AgBiBr6 thin films. J. Mater. Chem. C. 6, 9513–9520. https://doi.org/10.1039/C8TC03190E (2018).
Benmoussa, A., Bessai, N., Djelloul, A. & Rached, D. Electronic structure and optical properties of lead-free Cs2AgBiX6 (X = Cl, Br) double perovskites: A DFT investigation. Heliyon 10, e39285. https://doi.org/10.1016/j.heliyon.2024.e39285 (2024).
Ghebouli, M. A., Bouferrache, K., Alanazi, F. K., Ghebouli, B. & Fatmi, M. Computational insights into the stability, mechanical, optoelectronic, and thermoelectric characteristics investigation on lead-based double perovskites of (Cs₂, K₂, Rb₂)PbCl₆. Adv. Theory Simul. 8, 2400938. https://doi.org/10.1002/adts.202400938 (2025).
Djemli, A. et al. Structural, elastic, mechanic, electronic, and thermodynamic of limon₂ compound for electronic and energy storage. Phys. Solid State. 67, 356–366. https://doi.org/10.1134/S106378342305005X (2025).
Benamrani, A. et al. Structural, elastic, electronic, thermoelectric, and thermodynamic properties of cubic lamgx₂ (X = Cd, zn, Hg): For sustainable technologies. Phys. Solid State. 15, 055109. https://doi.org/10.1134/S1063783425050096 (2025).
Bioud, N. et al. Computational investigation of thermodynamic and mechanical properties of B2-type CoTi intermetallic compound. Phys. Solid State. 67, 68–74. https://doi.org/10.1134/S1063783425010084 (2025).
Ghebouli, M. A., Ghebouli, B., Chihi, T. & Fatmi, M. Predicted structural, elastic, electronic, lattice dynamic, thermodynamic and optical properties of cubic scf3 from first-principles calculations. Russ J. Phys. Chem. A. 95, S296–S306. https://doi.org/10.1134/S003602442102008X (2021).
Kumar, N. R. & Radhakrishnan, R. Electronic, optical and mechanical properties of lead-free halide double perovskites using first-principles density functional theory. Mater. Lett. 227, 289–291. https://doi.org/10.1016/j.matlet.2018.05.074 (2018).
Chen, P., Huang, Y., Shi, Z., Chen, X. & Li, N. Improving the catalytic CO2 reduction on Cs2AgBiBr6 by halide defect engineering: A DFT study. Materials 14, 2469. https://doi.org/10.3390/ma14102469 (2021).
Sotelo-Lerma, M. et al. CsPbBr₃ and Cs2AgBiBr6 composite Thick films with potential photodetector applications. Materials 17, 5123. https://doi.org/10.3390/ma17205123 (2024).
Thawarkar, S., Rondiya, S. R., Dzade, N. Y., Khupse, N. & Jadkar, S. Experimental and theoretical investigation of the structural and optoelectronic properties of Fe-doped lead-free Cs2AgBiCl6 double perovskite. Chem. Eur. J. 27, 7408–7417. https://doi.org/10.1002/chem.202100927 (2021).
Bouferrache, K. et al. Multi-property characterization of emerging functional materials. Bull. Mater. Sci. 47, 102. https://doi.org/10.1007/s12034-024-03291-3 (2024).
Wanniarachchi, W. A. C. P. et al. Cs2AgBiBr6 as a mixed anion perovskite for photovoltaic applications: A first-principles study. Mater. Today Proc. 64, 1783–1788. https://doi.org/10.1016/j.matpr.2022.03.012 (2022).
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University, Arar, KSA for funding this research work through the project number NBU-FFR-2025-310-15.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.A. Ghebouli, Formal analysis: K. Bouferrache, B. GhebouliMethodology: M.A. Ghebouli Writing and review & editing: R. BoudissaValidation : M. Fatmi, Faisal Katib Alanazi.
Corresponding author
Ethics declarations
Conflicts of interest
NO potential conflicts of interest of this article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ghebouli, M.A., Bouferrache, K., Boudissa, R. et al. Exploring the mechanical, dynamical, and thermal stability of Cs2AgBiX6 (X = Br, Cl) for optoelectronic and thermoelectric applications. Sci Rep 15, 20993 (2025). https://doi.org/10.1038/s41598-025-05549-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-05549-1
Keywords
This article is cited by
-
Effect of halogen substitution on the electronic and optical behavior of C₁₆H₁₀X₂O₂(X = F, cl, Br and I) organic semiconductors
Scientific Reports (2025)
-
DFT Investigation of Physical Properties of the Double Perovskites Cs2CaMCl6 (M = Ge or Pb)
Transactions on Electrical and Electronic Materials (2025)
