
Abstract
Industrial development has caused significant environmental pollution by discharging chromium (Cr) contaminated hazardous effluents into the ecosystem. The main goal of this study was to isolate, characterize, and assess the Cr(VI) resistance potential of indigenous naturally occurring chromate-resistant bacteria from the Uttar Dinajpur region of West Bengal. Based on the maximum tolerable concentration (MTC) to Cr(VI), a potent indigenous bacterial isolate with an MTC of 400 µg mL− 1 was selected for a detailed assessment of its Cr(VI) tolerance and reducing abilities. Molecular identification and phylogenetics revealed the isolate as a strain of Bacillus wiedmannii. Cr(VI) resistance ability of the isolate in different pH was analyzed, and a Fourier-transform infrared spectroscopy analysis under different pH in the presence of Cr(VI) was performed to capture the preliminary variation in the metabolic fingerprints. The isolate demonstrated Cr(VI) removal efficiency of 70.27% Cr(VI) at pH 8. The expression of chromate reductase enzyme in constitutive and Cr(VI) induced conditions was measured using different electron donors, and a detailed statistical analysis was performed to determine whether there exists a significant difference in the specific activity of chromate reductase in constitutive and induced conditions. The extracellular chromate reductase had a 98.42% Cr(VI) reduction rate using glucose as an electron donor and it was significantly higher than that of other electron donors, indicating glucose as the preferred electron source for Cr(VI) reduction. The genes responsible for coding enzymes responsible for chromate reduction viz., azoreductase, FMN-dependent NADH-azoreductase type1, and chromate reductase were also detected in the studied isolate. The findings of this study may contribute to the pool of indigenous isolates in countering chromate toxicity and their efficient utilization in chromate detoxification and bioremediation.
Similar content being viewed by others
Introduction
Heavy metal-resistant bacteria play a significant role in the biogeochemical cycling of metals, and bioremediation techniques have used the intrinsic detoxifying mechanisms of bacteria that facilitate the alteration of toxic metals into less harmful forms1,2. Bacteria utilize five primary strategies to mitigate the toxic effects of heavy metals. These include extracellular and intracellular sequestration of metal ions, formation of extracellular barrier, active transport or efflux of metal ions, reduction of toxic metal ions, and the biomethylation of metal ions3,4. Among all the recalcitrant heavy metals, chromium (Cr) is the 21st most abundant metal in the lithosphere, creating severe adverse effects on biota5. Cr may prevail in different oxidation states in an ecosystem due to its strong redox potential6. However, among the different oxidation states of Cr, hexavalent Cr or Cr(VI) mainly exists in nature due to various anthropogenic activities such as agricultural practices, mining, combustion of fossil fuels, painting, and plastic industries, and discharge of untreated wastewater and effluents from tanneries in the ecosystem7,8,9. In terms of cytotoxicity, Cr(VI) is a hundred times more toxic than Cr(III)10. Cr(VI) has several remarkable features, like strong oxidation potential, prompt permeability through biological membranes, and higher solubility in water, and the existence of such attributes is the primary reason to consider it a noxious element11. At elevated concentrations, Cr has the potential to cause mutations at molecular and cellular levels, but at trace amounts, it is a necessary nutritional element for the living domain12.
Microorganisms employ various strategies to endure heavy metal toxicity such as biosorption, bioaccumulation, and biotransformation13,14. Biosorption is a physicochemical mechanism where metal pollutants adhere to the surface of dead, inactive, or living biomass. This process represents a natural capability of specific biomass, allowing contaminants to bond chemically with the cell surface14. In contrast, bioaccumulation is a metabolically active process that is driven by the respiratory energy of living organisms. During such a process, Cr(VI) is incorporated into the living cell wall14. An alternative method for reducing the concentration of Cr(VI) is biotransformation, a process in which Cr(VI) is transformed to Cr(III) by the action of microorganisms14. Reports are available on the biotransformation of toxic Cr(VI) into less toxic Cr(III)15,16 by the pure and enriched mixed cultures of chromate-reducing bacteria (CRB). Many environmental factors involved in the transformation and bioavailability of Cr, including pH and temperature, are critical in altering the Cr(VI) detoxification competence of bacteria17.
In bacteria, chromate-reducing enzymes or chromate reductase are present in membrane-bound and cytosolic forms. Due to the presence of a soluble chromate reductase enzyme in the cellular system, CRB can reduce Cr(VI) more efficiently18,19. There are two kinds of enzymes that are engaged in the reduction of Cr(VI): class I chromate reductases are only involved in the reduction of Cr(VI), whereas class II chromate reductases are involved in other pathways either directly or indirectly20. Studies have reported the presence of different chromate reductases such as ChrR, YieF, NemA, LpDH, AzoR, and YhdA in soluble fractions or bound to the bacterial cell membrane21,22. It has been reported that conventional methods are more complicated for detoxifying (VI) containing waste mixtures from the environment. Therefore, it is essential to explore the indigenous CRB-based microbial remediation methods to protect the ecosystems from Cr(VI) contamination12,23,24.
An attempt was made in this study to identify potent indigenous Cr (VI) resistant bacteria from the sludge of a polluted pond in Raiganj, a town in the Uttar Dinajpur district of West Bengal, India. The present study was designed to achieve the following objectives- isolation of a potent and effective bacterial strain capable of tolerating elevated concentrations of Cr (VI), metabolic fingerprinting of the strain grown in the presence of Cr(VI) to assess the compositional changes in bacterial cell surfaces due to molecular binding with Cr, assessment of Cr(VI) removal efficiency and capacity of the isolate, estimation of chromate reductase enzyme activity in constitutive and Cr(VI)-induced conditions using different electron donors such as NADH, glutathione (GSH), and D-glucose, detailed statistical analysis for the determination of any significant difference in the specific activity of chromate reductase in the constitutive and induced conditions and in the presence of different electron donors, identification of the different genes encoding various chromate-reducing enzymes harboured within the genome of the bacterium and comprehend the mechanism of Cr(VI) detoxification. The findings of this study can help contribute to designing strategies for bioremediation of Cr (VI)-contaminated wastes by indigenous bacteria and also to the existing knowledge base on bacterial Cr(VI) detoxification.
Materials and methods
Isolation and screening of Cr(VI) resistant bacteria
Sludge samples collected from the polluted pond were subjected to concomitant isolation and screening to obtain Cr(VI) resistant bacterial strains. About 2 g of sludge from each sample was mixed with 20 mL of sterile double distilled water in a 100 mL sterile Erlenmeyer flask and properly mixed using a vortexer for 10–15 min. The obtained stock solution was subjected to serial dilution (up to 10− 4). Following the standard pour plate technique, 100 µL of each serially diluted sludge sample was poured aseptically in Petri plates containing 20 mL of tryptone soya agar (TSA) medium (pH 7.3 ± 0.2) supplemented with 200 µg mL− 1 of Cr(VI) and incubated at 35oC for at least 96 h25. The tolerant bacterial colonies obtained on the culture plates were repeatedly separated by the streak plate method and transferred and maintained in TSA slants.
Detection and estimation of total cr content from sludge sample
The obtained sludge sample was subjected to inductively coupled plasma atomic emission spectroscopy (ICP-AES) for Cr detection and quantitative analysis. To estimate the total Cr content of the sample, firstly, 0.5 g of sludge sample was taken in a Teflon beaker, and gradually, 10 mL of nitric acid and 5 mL of hydrofluoric acid were added for digestion. Then, the digested sample was heated on a hot plate. Next, 10 mL of aqua-regia was added to the dried sample mass, followed by reheating until everything dissolved, and the final volume of the solution was made up to 100 mL with sterile double distilled water. Finally, the prepared solution was run through the ARCOS, simultaneous ICP Spectrometer (SPECTRO Analytical Instruments GmbH, Germany).
Determination of maximum tolerable concentration (MTC) of Cr(VI)
The highest Cr(VI) concentration in the culture medium, non-lethal to the test organisms, was designated the MTC. The obtained bacterial isolates were initially grown in a sterile tryptone soya broth (TSB) medium (pH 7.3 ± 0.2). After 24 h of incubation at 35oC with shaking at 120 rpm, aliquots of 100 µL of the bacterial culture were inoculated in Cr-supplemented TSB media in 100 mL Erlenmeyer flasks with the Cr(VI) concentration ranging from 200 µg mL− 1 to 1000 µg mL− 1. A 10 mg mL− 1 Cr(VI) stock solution was prepared by dissolving potassium dichromate salt (K2Cr2O7) in deionized water and diluted to obtain working concentrations ranging from 200 µg mL− 1 to 1000 µg mL− 1. All the flasks were incubated at 35oC at 120 rpm for 96 h in an orbital shaking incubator (model SVIS-301 M). The bacterial growth in Cr(VI) containing media was monitored up to 96 h spectrophotometrically using a UV-visible spectrophotometer (Analytical Technologies, model UV-2080T) at 600 nm wavelength, and the optical density (O. D.) values were recorded.
Molecular identification and phylogenetic analysis of potent Cr(VI) resistant bacteria
The potent Cr(VI) resistant isolate was identified using 16S rRNA gene sequencing. Firstly, genomic DNA was isolated from the single bacterial colony using the standard phenol-chloroform method26. After that, the quality of DNA was evaluated on 1.0% agarose gel, followed by PCR amplification of 16S rRNA gene fragment using 27F [5’-AGAGTTTGATCCTGGCTCAG-3’] and 1492R [5’-CGGTTACCTTGTTACGACTT-3’] universal primers27. The PCR amplicons were purified by PEG-NaCl precipitation to remove contaminants. After that, the amplified PCR products were sequenced using a BDT V3.1 Cycle sequencing kit on an ABI 3730XL Genetic Analyzer. Sequence assembly was performed using BioEdit 7.2 software28. The assembled 16S rRNA gene sequence was deposited in the GenBank repository bearing accession number PQ567212. After sequencing, the acquired 16S rRNA gene sequence was used to carry out nucleotide BLAST against the 16S rRNA gene sequences database containing curated type strain sequences from bacteria and archaea29,30. The List of Prokaryotic Names with Standing in Nomenclature (LPSN) (accessible at https://lpsn.dsmz.de) was used to validate the type strain sequences that had the highest degree of sequence similarity with the 16S rRNA gene sequence of the studied isolate31,32.
A molecular phylogenetic analysis was performed to corroborate the isolate’s identity and homology with other closely allied intrageneric-type strains. To infer phylogeny, a comprehensive molecular phylogenetic analysis was carried out using the 16S rRNA gene sequence of the obtained strain (SA1) with the other closely related type strains detected using nucleotide BLAST30. Then, the 16S rRNA gene sequence of SA1 and other selected type strains were used to perform a multiple sequence alignment (MSA) using Clustal Omega33. The maximum likelihood (ML) approach was used to infer phylogeny following a comprehensive model test using MEGA 1134,35. The evolutionary model with the lowest Bayesian Information Criterion (BIC) value was applied to infer phylogeny using 1000 bootstrap replicates.
Morphological characterization
Overnight grown culture of the selected isolate was utilized for Gram staining using a staining kit (Himedia, India). A compound microscope with bright field illumination (Olympus, CH20i) was used to observe the cellular structure of the isolate28. For a comprehensive analysis of cellular structure, scanning electron microscopy (SEM) was performed using 24 h old bacterial culture. For this, cell masses were fixed in a 2.5% glutaraldehyde solution and then washed in sodium phosphate buffer (pH 7.2) twice. Graded ethanol series (20–100%) were used for dehydration, followed by gold sputter coating of the specimen using a sputter coater (Quorum Technologies, model SC7620). The dried samples were scanned using an environmental scanning electron microscope (Carl Zeiss, EVO18, ver. 6.02).
Study of bacterial growth kinetics in control and Cr(VI) stressed condition
The potent isolate’s growth kinetics was studied in the presence and absence of Cr(VI) in the TSB medium. Measurable growth was observed in the culture medium supplemented with Cr(VI) at the MTC of 400 µg mL− 1. The respective O.D. values were measured using a UV-visible spectrophotometer (Analytical Technologies, model UV-2080T) at 600 nm wavelength at every 2 h interval and were plotted graphically against time, and the growth rate was estimated.
Effect of pH on Cr(VI) resistance
The isolate was cultured in minimal salts medium (MSM) supplemented with Cr(VI) at the MTC (400 µg mL− 1) with pH varying from 4 to 12 to assess its Cr(VI) resistance capacity in different pH. The MSM comprised of Na2HPO4 4.5 g L− 1, KH2PO4 1.5 g L− 1, NH4Cl 0.3 g L− 1, MgSO4 0.1 g L− 1, yeast extract 0.5 g L− 1 and glucose 5 g L− 1. The glucose solution was sterilized using a 0.22 μm nylon membrane syringe filter and added aseptically into the other sterilized ingredients of the MSM. Media pH was adjusted using 0.1 N HCl or 1.0 M NaOH, and finally, the pH was measured with a pH meter (Labtronics, CE LT-50). About 0.1 mL of freshly revived bacterial culture was aseptically inoculated in 20 mL of MSM containing Erlenmeyer flasks with different pH, and it was incubated in a shaking incubator (model SVIS-301 M) at 35oC for 72 h with shaking at 120 rpm. Two control sets were also prepared. In the positive control setup, the isolate was grown at variable pH without Cr(VI) supplementation. In the negative control setup, the medium was supplemented with Cr(VI) without bacterium. Each experiment was carried out in triplicates, and the bacterial growth was then determined spectrophotometrically (Analytical Technologies, model UV-2080T) at 600 nm at every 2 h interval, plotted graphically against time, and the growth rate was estimated.
FTIR spectroscopy-based metabolic profiling
When analyzing structural and compositional changes in bacterial cell surfaces due to molecular binding with heavy metal ions, FTIR spectroscopy is an effective, reliable, and quick analytical method that offers information on the nature of heavy metal interaction36,37. FTIR spectroscopy was carried out to elucidate the effect of acidic, neutral, and alkaline pH in the presence and absence of Cr(VI) on the expression of functional groups present on the bacterial cell surface. Initially, the bacterial cell masses were grown in TSB media containing 400 µg mL− 1 Cr(VI) at different pH (pH 6, pH 7, and pH 11) separately, including the control (i.e., without metal) at 35oC. Due to the induced metal stress, some of the functional groups in the bacterial cell surface may be altered. Hence, FTIR-based spectroscopic profiling was employed to detect these changes. After 24 h of incubation, the cell pellets from each experimental set were harvested in 100 mL centrifuge tubes (10000 rpm for 20 min) by centrifugation (REMI C-24 Plus). In a hot air oven, these pellets were dried at 45–50oC for 24 h after washing with 0.5% NaCl solution thrice and crushed in an agate mortar and pestle. 0.05 g KBr was added to each resulting dry mass and pellets were cast. After that, an FTIR spectroscopic scan was carried out on a Vertex 80 FTIR system (Bruker, Germany), and each sample was scanned 64 times. The OPUS 8.2 software was used for data processing. The spectral data were collected over the 450–4000 cm− 1 range with 4 cm− 1 spectral resolution and baseline correction38.
Estimation of Cr(VI) removal capacity and efficiency
The bacterial isolate was cultured in 50 mL of freshly prepared sterile MSM supplemented with 200 µg mL− 1 of Cr(VI) at different pH (pH 7 and 8) separately to obtain viable cell mass for bioaccumulation. After that, 0.1 mL of freshly revived aliquot was aseptically inoculated in sterile MSM supplemented with 200 µg mL− 1 of Cr(VI) at different pH, and it was incubated in an orbital shaking incubator at 35oC with shaking at 120 rpm until the culture growth reached the mid-log phase. After that, the cell pellets from each experimental set were harvested in 100 mL centrifuge tubes by centrifugation (10000 rpm for 20 min at 4⁰C) using a centrifuge (REMI C-24 Plus), and the culture supernatant was collected for acid digestion using ultrapure nitric acid (65% v/v). The obtained cell pellets from each experimental setup were repeatedly washed with 1X phosphate buffer saline (PBS) solution (pH 7.4). These pellets were then dried in a hot air oven at 45–50oC. The final dried cell mass was measured and used to determine the isolate’s Cr(VI) removal capacity and Cr(VI) removal efficiency. A set of negative controls was prepared where the medium contained no bacterial inoculum to determine the amount of passive Cr(VI) uptake by the medium constituents. This was done for background correction so that the Cr(VI) uptake by only the viable bacterial cells could be detected and not due to the interaction between the MSM constituents and Cr(VI). The initial and residual Cr(VI) concentrations in all the culture media were measured using inductively coupled plasma optical emission spectroscopy (Perkin Elmer Optima 5300 DV). The metal removal efficiency and capacity of the isolate were calculated using the following equations39:
where Ci represents the initial concentration of Cr(VI) (mg L− 1) in solution, Cf represents the residual concentration of Cr(VI) (mg L− 1) in solution, V represents the volume of the solution in L, and M represents the dry weight of bacterial biomass in g.
Analysis of chromate reducing activity
To assess the chromate reducing activity of the potent isolate, freshly revived 0.1 mL of bacterial inoculum was aseptically transferred into the sterile MSM (pH 7.2 ± 0.2), and it was incubated in a rotary shaker incubator at 35oC for 20 h (mid-log phase of growth). After incubation, the cell masses were separated using a centrifuge (RPM = 10000 for 10 min at 4oC), and the culture supernatant was collected as a source of chromate-reducing enzymes18. The constitutive expression (no chromate in culture medium) of chromate reductase activity of the culture supernatant (extracellular enzyme extract) was assayed using three different electron donors, viz., reduced glutathione (GSH), NADH, and D-glucose18,40.
The assay mixture (3 mL) contained 600 µL of 0.2 mM L− 1 Cr(VI) solution, 1200 µL of culture supernatant (as extracellular enzyme source), 600 µL of 200 mM L− 1 phosphate buffer (pH 7.2), and 600 µL of 20 mM L− 1 of any of the given electron donor (NADH or GSH or D-glucose solution). This reaction mixture was then incubated at 37oC for 30 min. After incubation, 1500 µL of 20% trichloroacetic acid was added to the assay mixture to stop the reaction. Finally, 6 mL of the complexing reagent 1,5-diphenylcarbazide/ethanol (0.5% w/v) was added to the assay mixture, and a pink hue was developed. The absorbance was determined spectrophotometrically at 540 nm wavelength18,41. A set of negative controls was prepared similarly without any bacterial culture supernatant as an enzyme source. The whole experiment was carried out thrice, each time using triplicates, and the mean and standard error were calculated. A standard curve representing the chromate concentration was also prepared. The Cr(VI) concentration was determined from the standard curve, and the reduced Cr(VI) concentration was used to calculate the enzyme activity. The amount of enzyme that reduced 1 µmol L− 1 of Cr(VI) per minute at 37oC was regarded as one unit (1 U) of chromate reductase activity40,41.
Estimation of chromate reductase activity in the induced condition
To perform this assay, the MSM (pH 7.2 ± 0.2) was prepared in the manner mentioned above, except an initial 25 µM, 50 µM and 100 µM concentration of Cr(VI) solution were added into the sterile medium (pH 7) after 10 h of bacterial growth (early log phase), and the culture supernatant was collected after 24 h (mid-log phase) incubation at 35ºC. The induced (presence of chromate in culture medium) expression of chromate reductase was estimated from the culture supernatant (extracellular enzyme) and expressed as U mg− 1. The whole experiment was carried out thrice, each time using triplicates, and the mean and standard error were calculated.
Estimation of total protein content and specific activity of chromate reductase
At the exponential growth phase, the cell masses grown in MSM (pH 7.2 ± 0.2) were harvested using a cooling centrifuge (REMI C-24 plus) in 15 mL centrifuge tubes (RPM = 10000 for 10 min at 4oC, and the cell-free supernatant was collected for total protein estimation. The total protein amount in the bacterium’s cell-free extract was assayed following the Lowry method42, and bovine serum albumin (BSA) was used for the standardization. The whole experiment was carried out thrice, each time using triplicates, and the mean and standard error were calculated. The specific activity of the chromate reductase enzyme was expressed as U mg− 1 and was calculated using the following formula:
Statistical analysis
Kruskal-Wallis one-way analysis of variance (ANOVA) on ranks was performed to detect statistically significant differences in specific activity of chromate reductase in the presence of various electron donors such as NADH, GSH, and D-glucose, both in constitutive and induced conditions. Following rank-based ANOVA, all pairwise comparisons and comparisons against a control group were performed using the Dunn’s test. Chromate reductase activity of the bacterium was induced in the culture using various Cr(VI) concentrations ranging from 25 µM to 100 µM. No Cr(VI) was supplemented in the culture medium to determine constitutive expression. Statistical analysis was carried out using SigmaPlot 12.0.
Detection and analysis of genes coding for chromate reductase
Genes encoding enzymes responsible for reducing chromate harboured by the genome of the potent isolate were explored by genomic DNA isolation and PCR amplification followed by sequencing. For genomic DNA isolation, the bacterium was grown overnight in an LB broth medium at 35ºC in an orbital shaking incubator. After overnight incubation, the fresh bacterial culture was harvested using centrifugation at 12000 rpm for 15 min at 4ºC (REMI C-24 Plus). According to the manufacturer’s protocol, the total genomic DNA from isolate SA1 was extracted using the Xploregen genomic DNA extraction buffer set. Subsequently, the electrophoresis of the obtained genomic DNA was carried out on 1.5% agarose gel (1X TBE, 5V/cm for 1 h). Whole genome sequences of different strains of Bacillus spp. were obtained from GenBank to mine genes encoding different chromate reductase enzymes, and Bacillus-specific PCR primers were designed using the NCBI Primer-BLAST tool. Bacillus-specific primer sequences used for the azoreductase gene were forward [5’-TTGGTAGAAGCTCGCGTGGTG-3’] and reverse [5’-CATAAACACCTCTAGCGACCGT-3’]. The primers for the FMN-dependent NADH-azoreductase gene were forward [5’-GCGGAGCAAGCAGTTAGTTC-3’] and reverse [5’-TGCCATTTCCATACGAGCCA-3’]. The primer sequences used for detecting the chrR gene (encoding chromate reductase) were forward [5’-TCCACCACGAATACGTGCGTGCAA-3’] and reverse [5’-GACAACAGCGGCATTGACTC-3’].
The PCR reaction mixture (50 µL) used was composed of 1µL of extracted DNA, 2 µL of forward primer, 2 µL of reverse primer, 4 µL of 2.5 mM dNTPs, 10 µL of 10X Taq DNA polymerase assay buffer, 1 µL of Taq DNA Polymerase Enzyme (3 U mL− 1) and 30 µL of nuclease-free water. The PCR cycling conditions consist of initial denaturation at 94ºC for 3 min, followed by subsequent 30 cycles comprising of denaturation at 94ºC for 1 min, annealing at 50ºC for 1 min, extension at 72ºC for 2 min, and final extension at 72ºC for 7 min. The purification of PCR amplicons was carried out using a QIAquick PCR purification kit (QIAGEN). DNA Sequencing of PCR products and amplification of primers were carried out using BDT ver. 3.1 cycle sequencing kit and nucleotide sequence analysis were performed using ABI 3130 Genetic Analyzer. After that, sequence assembly, alignment, and contig generation were carried out using BioEdit 7.2. The assembled gene sequences were used to perform translated BLAST (tblastx) to find their similarity with the genes encoding chromate reductases. InterProScan facility by InterPro v95.0 (https://www.ebi.ac.uk/interpro/) was used for the prediction of domains, classification of the enzymes, and functional validation43. The acquired gene sequences were deposited in GenBank, and accession numbers were obtained (PV056065 to PV056067).
Results
Isolation and screening of Cr(VI) resistant bacteria
Based on colony morphology, initially, three Cr(VI) resistant bacterial isolates were obtained. The isolate SA1 exhibited comparatively vigorous growth in culture media. The colonies of SA1 appeared as moist, shiny, and moderately white with irregular, undulate peripheral edges and were slightly elevated on the agar surface (supplementary Fig. S1).
Detection and estimation of total cr in sludge
The presence of Cr was detected in the sludge sample using ICP-AES. The estimated total Cr concentration was 2.78 mg kg− 1 of sludge.
MTC of Cr(VI) of the isolates
The isolate SA1 depicted comparatively higher MTC, i.e., 400 µg mL− 1, than the other two isolates, RA4 and RA5, where the MTC for Cr(VI) was only 100 µg mL− 1 (supplementary Table S1). As the isolate SA1 showed relatively higher MTC to Cr(VI), it was thoroughly characterized and studied to assess its Cr(VI) resistance and reducing capacity and mechanisms of Cr(VI) resistance.
Molecular identification and phylogenetic analysis of potent Cr(VI) resistant bacteria
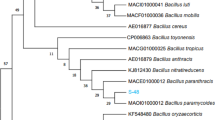
Nucleotide BLAST of the 16S rRNA gene sequence of SA1 (GenBank accession number PQ567212) revealed 99.5% similarity with Bacillus wiedmannii FSL W8-0169T type strain. The ML method with the Hasegawa-Kishino-Yano substitution model was used to infer the molecular phylogeny as it demonstrated the lowest Bayesian Information Criterion (BIC) value. Based on the molecular phylogenetic analysis, the studied isolate SA1 exhibited proximity with the type strain Bacillus wiedmannii FSL W8-0169T and was found to reside in the same clade, sharing the most common ancestor. The phylogenetic tree of SA1 is depicted in Fig. 1.

A circular phylogenetic tree of SA1 constructed using 16S rRNA gene sequences of closely related valid type strains of Bacillus spp.The Streptococcus acidominimus DSM 20622 type strain was used as an outgroup. The evolutionary history was inferred using the Maximum Likelihood method and Hasegawa-Kishino-Yano model using 1000 bootstrap replicates.
Morphological characterization
Gram staining confirmed the isolate as a rod-shaped Gram-positive bacterium. Utilizing SEM, the length of each rod was found to range between 3 and 5 μm. The SEM micrograph of the isolate along with cellular measurements is shown in supplementary Fig. S2.
Comparative growth kinetics in the presence and absence of Cr(VI)
The growth of SA1 was relatively abundant in the control set, i.e., without Cr(VI), when compared to the Cr(VI) supplemented culture medium. The growth rate of SA1 in the control setup was estimated at 3.0 generations h− 1 with a generation time of about 20 min. Bacterial growth in the presence of Cr(VI) displayed a recognizable pattern with a preliminary extended lag phase followed by subsequent exponential and stationary phases. The subsequent growth rate and generation time of SA1 in the presence of Cr(VI) was 1.47 generations h− 1 and 40.7 min, respectively. Figure 2 shows the growth curves of the bacterial isolate both in the presence and absence of Cr(VI).

Growth pattern of the isolate SA1 in both control TSB medium and 400 µgm L− 1 (i.e., the MTC value of the isolate at 35ºC) Cr(VI) supplemented TSB medium. The error bars represent the standard deviation around the mean.
Effect of pH on Cr(VI) resistance
In the control set, i.e., without Cr(VI) supplementation, SA1 exhibited growth at pH 6, 7, 8, 9, 10, and 11. In the presence of 400 µg mL− 1 Cr(VI), visible growth was evident from pH 6 to 11, but the growth was relatively less, as determined from the O.D. values. The results showed that in the control set of pH 6, 7, 8, 9, 10, and 11, the bacterial growth exhibited a relatively shorter lag phase of about 1–2 h. Growth in the Cr(VI) supplemented MSM at pH 6, 10, and 11 depicted extended lag phases of 6 h, 4 h, and 12 h, respectively. In Cr(VI) supplemented MSM at pH 7, 8, and 9, the lag phase was relatively shorter (1–2 h). Bacterial growth was completely absent in MSM with pH 4, 5, and 12 both in the absence and presence of 400 µg mL− 1 Cr(VI). SA1 demonstrated the highest growth rate in pH 7 both in the absence (3.0 generations h− 1) and presence (1.47 generations h− 1) of Cr (VI). However, the growth rate was minimal at pH 11 in the absence (0.36 generations h− 1) and the presence (0.24 generations h− 1) of Cr(VI). The growth rate and generation time of SA1 at different pH, both in the control condition and in Cr(VI) supplemented MSM, is shown in supplementary Table S2. The results further suggested that the growth of the isolate SA1 was optimal in neutral to slightly alkaline pH. This was true for the control and Cr(VI) supplemented conditions. Since the isolate demonstrated optimal growth in pH 7, further analysis of chromate reductase activity by SA1 was performed at neutral pH.
Comparative metabolic profiling under cumulative Cr(VI) and pH stress
The metabolic profiling revealed differential FTIR spectra indicating the presence and absence of varied functional groups at different pH under constant Cr(VI) stress of 400 µg mL− 1 at 35ºC. In the control set at pH 6 (without Cr(VI) stress), the spectrum showed sharp peaks at 3430.11 cm− 1 and 2925.42 cm− 1 region as a result of the stretching of the N-H bond of amino groups along with the presence of γO-H of hydroxyl groups and the asymmetric stretching vibration of the γc-H bond of the –CH2 groups integrated with that of the –CH3 groups, respectively44. Similarly, at pH 6 supplemented with Cr(VI) at 400 µg mL− 1, it showed peaks at 3447.58 cm− 1 and 2923.41 cm− 1. A single sharp peak was detected at 1634.81 cm− 1 and 1655.15 cm− 1 spectral regions in the control and metal-treated set of pH 6, respectively. This was probably due to the presence of C = O group vibrations in the peptide bond, and these absorbance bands may be seen in amino sugars from cell-associated polysaccharides with N-acetyl/glucuronamide groups44,45. A sharp peak appeared in the 1405.73 cm− 1 region of the control set (pH 6) due to carboxyl group vibration. At the same time, a shift to 1385.21 cm− 1 lower energy level was noticed in the Cr(VI) loaded sample (pH 6) as a consequence of symmetric stretching of COO- vibration44. Owing to the existence of the carboxyl (-COOH) group and phosphate group (P = O and P-O of the C-PO32− moiety) vibration, sharp peaks originated in 1283.74 cm− 1, 1078.02 cm− 1, 1058.03 cm− 1 and 1077.76 cm− 1 spectral region of control and metal treated sets (pH 6), respectively44. In the control set (pH 7), sharp broad-based peaks appeared in 3430.22 cm− 1 and 2922.84 cm− 1 region, whereas 3432.59 cm− 1 and 2922.84 cm− 1 spectral region for Cr(VI) exposed samples. A minor change was observed in the spectral region of the metal-loaded set. A single sharp peak was found in the 1635.85 cm− 1 and 1637.82 cm− 1 regions of the control and metal-loaded samples, respectively. Other complex peaks were found at 1405.16 cm− 1, 1077.66 cm− 1, 1403.75 cm− 1, and 1075.76 cm− 1 region for control and metal accumulated samples. The spectrum of the control set (pH 11) showed a characteristic peak at 3410.82 cm− 1 region, and a sharp peak was found at 3431.08 cm− 1 region for the metal-loaded sample (pH 11). Other sharp peaks were noticed at 2924.72 cm− 1, 1634.72 cm− 1, 1406.65 cm− 1, and 1044.50 cm− 1 spectral region for the pH 11 control set, whereas 2923.59 cm− 1, 1638.49 cm− 1, 1384.60 cm− 1, and 1059.63 cm− 1 region for pH 11 with Cr(VI) (400 µg mL− 1) loaded sample. One of the important observations of the overall investigation was the appearance of peaks at 1385.21 cm− 1 and 1384.60 cm− 1 region for acidic and basic metal-treated samples because of symmetric stretching of COO- group vibration. Another interesting observation was the appearance of a peak at 1283.74 cm− 1 wave number for the control set at pH 6. The supplementary Table S3 represents the major absorption peaks observed in FTIR spectra of the bacterium in response to pH and Cr(VI) (400 µg mL− 1) stress and probable functional groups involved in metal binding. It appeared due to carboxyl and phosphate groups’ vibration in cell-associated polysaccharides, peptides, peptidoglycan, and phospholipids44. Fig. 3 represents the comparative FTIR spectra under different pH (i.e., pH 6, 7, and 11) as well as in response to different pH (i.e., pH 6, 7, and 11) with constant Cr(VI) (400 µg mL− 1) stress at 35ºC.

A stacked FTIR spectra plot of SA1 at different pH (pH 6, 7and 11) in the control condition and when exposed to Cr(VI) at 400 µg mL− 1 stress at 35ºC.
Cr(VI) removal capacity and efficiency
The metal removal efficiency and metal removal capacity of isolate SA1 were estimated when exposed to 200 µg ml− 1 of Cr(VI) supplemented medium at different pH (pH 7 and pH 8) for 24 h. The Cr(VI) removal efficiency of the isolate SA1 was measured be 70.27% at pH 8 and 69.31% at pH 7, while the metal removal capacity by the isolate was 141.74 ± 0.565 mg g− 1 of Cr(VI) at pH 8 and 139.8 ± 0.611 mg g− 1 of Cr(VI) at pH 7. The concentration of Cr(VI) remained unchanged in the abiotic control setup of pH 7 and pH 8. The Cr(VI) concentration in abiotic control was estimated to be 201.7 ± 1.761 µg ml− 1 at pH 7 and 201.7 ± 1.871 µg ml− 1 at pH 8.
Comparative chromate reduction (constitutive expression) using different electron donors
A statistical analysis using Kruskal-Wallis ANOVA on ranks was performed to determine whether there is any significant difference in the specific activity of chromate reductase (when constitutively expressed) in the presence of different electron donors like NADH, GSH, and D-glucose. The results demonstrated a statistically significant difference (H = 23.228,2 d. f., p < 0.01). Pairwise multiple comparison tests using Dunn’s method depicted significant differences (p < 0.01) in the specific activity of chromate reductase between GSH and NADH (Q = 2.405) and GSH and glucose as electron donors (Q = 4.811). The specific activity was also significantly different when NADH was replaced with D-glucose as the electron donor (Q = 2.405). A graph showing the comparative specific activities of constitutively expressed chromate reductase in the presence of three different electron donors is depicted in Fig. 4.

A graph showing the comparative specific activities of constitutively (0 µM) and inducibly expressed chromate reductase (induced with 25 µM, 50 µM, and 100 µM chromate) in the presence of three different electron donors (NADH, GSH, and D-glucose). The asterisks denote statistically significant differences (increasing number of asterisks denotes increased significance) in chromate reductase specific activities at different conditions (connected by blue lines). The error bars represent the standard error of the mean.
Comparative chromate reductase expression (constitutive versus induced) using different electron donors
Kruskal-Wallis ANOVA on ranks was also performed to detect significant differences between the specific activity of chromate reductase when expressed constitutively and when induced using different concentrations of Cr(VI) under the influence of each type of electron donor separately. In the presence of NADH as an electron donor, a significant difference (H = 32.978, 3 d. f., p < 0.01) was detected in the specific activity of constitutively expressed chromate reductase with that of induced chromate reductase (induced using 25, 50 and 100 µM of Cr(VI)). Similar results were also observed when GSH (H = 30.004, p < 0.01) and D-glucose (H = 32.901, p < 0.01) were used as the electron donor. To detect the inducing concentrations of Cr(VI) that contribute to the difference in specific activity of chromate reductase with the constitutively expressed one, pairwise multiple comparison tests using Dunn’s method (p < 0.01) were carried out. Considering all three electron donors, the specific activity of constitutively expressed chromate reductase differed significantly from the specific activity of chromate reductase induced with 50 and 100 µM of Cr(VI). When NADH (Q = 1.812) and D-glucose (Q = 1.812) were used as electron donors, no significant difference in the specific activity of chromate reductase was observed between constitutively expressed and the one induced with 25 µM of Cr(VI). However, when GSH (Q = 3.020) acted as an electron donor, a significant difference was observed between the specific activity of chromate reductase expressed constitutively and induced with 25 µM of Cr(VI).
Comparative chromate reduction (induced expression) using different electron donors
A statistical analysis using Kruskal-Wallis ANOVA on ranks was performed to determine further whether there is any significant difference in the specific activity of chromate reductase when induced with different concentrations of Cr(VI), i.e., 25, 50, and 100 µM using either of the three electron donors separately.
In the presence of NADH and D-glucose, pairwise multiple comparison tests detected a statistically significant difference in chromate reductase specific activity when induced with different Cr(VI) concentrations, i.e., 25, 50, and 100 µM. However, when GSH was used as the source of electrons, the specific activity showed no significant difference between the bacteria-induced with 25 and 100 µM of Cr(VI).
A graph showing comparative specific activities of chromate reductase when induced with 25 µM, 50 µM, and 100 µM chromate concentration is depicted in Fig. 4.
Comparative analysis of induced chromate reductase expression using three different electron donors
Kruskal-Wallis ANOVA on ranks was also performed to capture the effect of various electron donors on the specific activity of chromate reductase, keeping the inducing concentration of Cr(VI) constant. For each inducing concentration, a statistically significant difference in the specific activity of chromate reductase was evident using different electron donors (H(25µM) = 23.214, H(50)µM = 19.344, H(100 µM) = 23.242). Pairwise multiple comparisons using Dunn’s test detected that when induced with 25 and 100 µM of Cr(VI), there is a significant difference in the chromate reductase specific activity between cells treated with different electron donors. For cells treated with 50 µM of Cr(VI), there was no significant difference between the specific activity of chromate reductase obtained from cells treated with GSH and D-glucose (Q = 1.336).
Analysis of genes encoding enzymes for chromate reduction
The assembled contigs obtained after amplifying (gel images of amplified PCR products given in supplementary Fig. S3) the selected genes responsible for Cr(VI) reduction were subjected to BLASTX (searching of protein databases using a translated nucleotide query). BLASTX demonstrated > 99% identity with different chromate reductase enzymes (FMN-dependent NADH-azoreductase, chromate reductase encoded by chrR and azoreductase). The contigs were also subjected to InterPro search (https://www.ebi.ac.uk/interpro/search/sequence/)46 and the results showed that the corresponding amino acid sequences of all the three contigs are part of the flavoprotein-like superfamily of proteins (IPR029039). The enzymes belonging to this protein superfamily catalyse the NAD(P)H-dependent two-electron reductions of quinones, protecting the cells from damage caused by free radicals and reactive oxygen species47. Chromate reductases are part of the FMN-dependent NADH: quinone oxidoreductase (azoreductase type 1) and NAD(P)H-dependent reductase protein family of the flavoprotein-like superfamily of proteins. The comprehensive details of the three contigs and their translated amino acid sequence counterparts are given in Table 1.
Discussion
Cr is a hazardous heavy metal with carcinogenic properties and wide industrial use. At high concentrations, it can lead to subsequent mutations on the cellular and molecular levels. From the bioremediation point of view, the reduction of Cr(VI) to Cr(III) by microorganisms is a predominant chromate toxicity elimination route22, and it is an essential technique for cleaning up Cr(VI) polluted sites48. In the present study, sludge samples were collected from a polluted pond in Raiganj town in the Uttar Dinajpur district of West Bengal. Three isolated bacterial strains showed Cr(VI) tolerance, but one isolate designated SA1 possessed a relatively higher MTC of 400 µg mL-1 to Cr(VI). This level of resistance notably surpasses the tolerance levels documented in prior studies on Bacillus sp. strains. It was reported that Bacillus sp. strain SFC500-1E displayed an MTC of 150 mg L-1 to Cr(VI)49, whereas Bacillus thuringiensis tolerated up to 520 µg mL-1 to Cr(VI)50. Another study reported that Bacillus sp. strain CRB-7 and Bacillus sp. strain CRB-B1 possess a higher tolerance of 450 mg L-1 and 420 mg L-1 to Cr(VI), respectively51,52. These comparisons highlight the relatively enhanced Cr(VI) tolerance capability of the isolate SA1 in the current study. Given its elevated tolerance, the isolate SA1 was selected for subsequent characterization to assess its potentiality for Cr(VI) reduction and removal, underscoring its potentiality for bioremediation of Cr(VI) contaminated environments. SEM analysis and Gram staining indicated that the studied isolate is a rod-shaped Gram-positive bacterium. Molecular identification using 16S rRNA gene sequencing confirmed the isolate as a strain of Bacillus wiedmannii. Bacillus strains exemplify indigenous microorganisms due to their ubiquitous distribution, evolutionary adaptation, and integral ecological functions within soil ecosystems. The genus Bacillus comprises Gram-positive, aerobic, or facultatively anaerobic, spore-forming bacteria that demonstrate remarkable environmental persistence and metabolic versatility53. The indigenous status ensures optimal survival and metabolic activity under site-specific conditions, making them superior candidates for in-situ bioremediation compared to allochthonous strains54. Their enzymatic capabilities enable the degradation of various pollutants including hydrocarbons, heavy metals, and xenobiotic compounds55.
The growth kinetics under Cr(VI) stress showed a relatively long lag phase followed by exponential growth. A prolonged lag phase during bacterial growth under metal stress conditions is the probable result of additional time the bacteria require to start physiological and metabolic changes, allowing them to grow and survive under metal stress56. The growth and metal stress resistance properties of bacterial strains are greatly influenced by pH57. Variation of pH has been identified as a crucial factor for chemical speciation, solubility, and bioavailability of Cr(VI)58. Studies have shown that pH significantly impacts bacterial Cr(VI) reduction, and the initial pH of the medium is vital for this purpose. The pH range between 7 and 8 is ideal for Bacillus sp.59. In the present study, the growth of the isolate was found to be different in varying pH conditions under 400 µg mL-1 Cr(VI) stress. In the presence of Cr(VI) stress, the bacterium showed visible growth between pH 6 to 11, but growth was absent in pH 4, 5, and 12. Analysis of metal stress resistance in different pH exhibited that the studied bacterium possesses the capacity to withstand a specific range of basic (i.e., pH 8–11) and acidic pH (i.e., pH 6) in the presence of Cr(VI)60. This finding suggests that basic and neutral or near-neutral pH are essential for Cr(VI) resistance. Some studies have identified Cr(VI) reducing bacterial species that can either reduce Cr(VI) at a pH close to neutral (7-7.8) or a pH higher than neutral61. Therefore, different bacterial cultures require optimal pH values for chromate bioremediation to maximize Cr(VI) reduction62.
Metabolic profiling of the studied isolate using FTIR suggested the presence of different functional groups, including O-H, C-O, C-H, COO-, C = C, P = O, and C = O. Analysis of spectral data depicted that, in the Cr(VI) stressed condition, changes in the absorption and intensities of the peaks were found due to the interaction between Cr(VI) and the functional groups in the bacterial cell surface44. Upon exposure to heavy metals, bacteria instinctively respond to the stress by modifying their biomolecular fingerprint and employing different survival mechanisms. This may involve an increase in the production and expression of specific cellular proteins and enzymes that either neutralize the toxic metals or block their entry into the cell28. In the present study, a mixture of distinct broad and sharp absorption peaks were observed at the different regions of the control and metal-loaded spectrum. The spectral band for the control set at pH 6 were shifted from 3430.11 cm− 1, 2925.42 cm− 1, 1634.81 cm− 1, 1405.73 cm− 1, 1283.74 cm− 1, 1078.02 cm− 1, and 1058.03 cm− 1 to 3447.58 cm− 1, 2923.41 cm− 1, 1655.15 cm− 1, 1385.21 cm− 1, and 1077.76 cm− 1 region, respectively after exposure to Cr(VI). This observation indicates the metal’s probable interaction with the bacterium’s cell surface protein. A minor alteration was detected in the spectral region of the control set and metal-loaded set at pH 7. The peaks for the control set at pH 7 were present around 3430.22 cm− 1, 2927.28 cm− 1, 1635.85 cm− 1, 1405.16 cm− 1, and 1077.66 cm− 1 spectral region. These peaks were shifted in the regions of 3432.59 cm− 1, 2922.84 cm− 1, 1637.82 cm− 1, 1403.75 cm− 1, and 1075.76 cm− 1 for the metal-loaded set at pH 7. For the control set at pH 11, the absorbance peaks appeared around 3410.82 cm− 1, 2924.72 cm− 1, 1634.72 cm− 1, 1406.65 cm− 1, and 1044.5 cm− 1 region. Post-exposure to Cr(VI) at pH 11, these spectral bands were shifted to 3431.08 cm− 1, 2923.59 cm− 1, 1638.49 cm− 1, 1384.6 cm− 1, and 1059.63 cm− 1, respectively. These spectral differences in both control and metal-loaded samples suggested that the distinct differences in the metabolic fingerprinting occur due to the Cr (VI) induced alterations at the molecular level of the bacterium’s cell surface protein. In the lipid region i.e., 3000 –2800 cm− 1 of the FTIR spectrum63, a minor variation was detected in the characteristics spectral bands of the control set (i.e., pH 6, pH 7, and pH 11) and metal-loaded set (i.e., pH 6, pH 7, and pH 11). However, in the protein region i.e., 1750 –1500 cm− 163, a combination of sharp and distinct absorption maxima was notably evident in the spectral region of the control set (i.e., pH 6, pH 7, and pH 11) and metal-loaded set (i.e., pH 6, pH 7, and pH 11). The characteristics of the absorption peaks in the combined region i.e., 1500 –1200 cm− 164 of the spectrum revealed a similar pattern for the control set at pH 6, pH 7, and pH 11, but the wavenumbers were different for each control set. On comparing the absorption spectrum, a similar pattern was also noticeable for the metal-loaded set at pH 6, pH 7, and pH 11 in the combined region of the spectrum. However, in the present study, distinct absorption peaks were also visible in the polysaccharide region i.e., 1200 –900 cm− 163 of the spectrum for the control set (i.e., pH 6, pH7, and pH 11) and metal-loaded set (i.e., pH 6, pH 7, and pH 11). These spectral regions delineate the characteristic signatures of various biomolecules localized on the bacterium’s cell surface28,36. Following Cr(VI) exposure, a gradual peak shift to the higher or lower wavelength indicates the peak’s weak and strong absorption. This result suggests the possible interaction of Cr(VI) with dominant functional groups of bacterial cell surface-associated proteins and polysaccharides. The results of this analysis were found to be in line with other studies28,44,65.
During recent years, different Cr(VI)-resistant Bacillus strains, including Bacillus cereus, Bacillus circulans, Bacillus firmus KUCr1, Bacillus megatarium TKW3, and Bacillus methylotrophicus have been isolated for Cr bioremediation purposes18,40,41,66,67,68,69,70,71. The ionization state of metal ions and different functional groups on the bacterial cell surface is influenced by the pH of the growth medium, which alters the total surface charge of the bacterial cell surface and impacts the effectiveness of microbial removal efficiency of heavy metals28,52. A further increase in the removal of metal by bacteria at alkaline pH may be caused by the deprotonation of metal binding sites in bacterial cell walls, which results from the culture media’s reduced proton content28,72. In this study, SA1 exhibited significant Cr(VI) reduction capabilities, achieving about 70% removal at pH 7 and 8 under an initial Cr(VI) concentration of 200 µg mL⁻¹. In comparison, previously reported Bacillus strains such as CRB-B1 demonstrated Cr(VI) removal efficiency of 86% over an extended incubation period of 48 h at 200 µg mL⁻¹ Cr(VI) concentrations and pH ranging from 6–852. A Bacillus paramycoides strain Cr6 has been shown to remove over 90% of Cr(VI) within 18 h at a Cr(VI) concentration of 200 mg L⁻¹, with optimal reduction observed at pH 8 and 31 °C73. Notably, isolate SA1 achieved substantial Cr(VI) removal within a shorter duration (24 h) at 35 °C, indicating its potential for rapid and effective bioremediation of Cr(VI)-contaminated environments. The efficacy of Cr(VI) removal by the cell mass of Bacillus was found to occur within a narrow pH range, suggesting its reliance on the pH of the medium. The findings of this analysis were found to be consistent with other previously reported studies52,74,75,76.
Some studies have reported that Bacillus sp. and other microorganisms, including Pseudomonas, Shewanella, and Enterobacter could reduce Cr(VI) by secreting specific soluble enzymes77,78. Microbes can synthesize enzymes extracellularly or intracellularly79. Bacterial chromate reductases are found in CRB’s membranous or cytosolic fractions80,81. Membrane-bound reductase enzymes are prominent in anaerobic CRB, whereas cytosolic soluble chromate reductases are found in aerobic CRB22. Soluble chromate reductases are significant for Cr(VI) bioremediation82. Some studies have reported that bacterial cells can export Cr(VI) reducing enzymes into the medium for reduction. These enzymes are extracellular60. Extracellular reduction of Cr(VI) is harmless for the bacterial cell because it has a lesser effect on DNA83. From the industrial and economic viewpoint, extracellular chromate reductase is helpful for easy downstream operations and is a cost-effective product18. This study detected high extracellular inducible chromate reductase activity in SA1. This study emphasizes the effect of three electron donors on constitutive and inducible enzyme expression and the impact of different Cr(VI) concentrations on inducible enzyme expression in the presence of various electron donors. A significant finding of the present study was that the specific activity of chromate reductase in the induced samples was higher than in the constitutive ones. Another significant finding of the present study was that maximum specific activity occurred at 30 min with 100 µM initial Cr(VI) supplementation (induced) using NADH and D-glucose as an electron donor, and the percentage of Cr(VI) reduction was found to be 95.20% and 98.42%, respectively in 30 min by the inducible extracellular chromate reductase enzyme. In the present study, an initial 50 µM Cr(VI) supplemented medium was optimum for the maximum specific activity (occurred at 30 min) using GSH as an electron donor and the percentage of Cr(VI) reduction was found to be 96.64% in 30 min by the inducible extracellular chromate reductase enzyme. Hence, an initial 50 µM and 100 µM Cr(VI) in the medium was optimum for the enzyme activity in the extracellular medium. This finding strongly indicates the presence of higher extracellular chromate reductase activity in the studied isolate, enabling the bacterium to cope with Cr(VI) stress. Earlier studies have suggested that extracellular chromate reductase activity is enhanced in the presence of NADH in Bacillus sp18,40. However, in the present study, the non-enzymatic reductant D-glucose demonstrated elevated chromate reduction activity. This finding is in line with earlier reports on Cr(VI) reduction by extracellular chromate reductase18,40,52,84,85,86,87. It was observed that SA1 preferred D-glucose as the electron donor for Cr(VI) bioreduction compared to NADH and GSH. A previous study reported that Bacillus sp. strain CRB-1 exhibited a significant Cr(VI) reduction efficiency of 76.64% in the presence of glucose. This study further revealed that the employment of glucose as an electron donor significantly enhances the microbial reduction of Cr(VI) highlighting its pivotal role in optimizing the bioremediation of Cr(VI)52. There is a lot of evidence of microbial Cr(VI) reduction using glucose as an electron donor85,86,87,88. Glucose is a relatively inexpensive, economically feasible and effective carbon source that can serve as an efficient electron donor for microbial redox processes. Glucose is metabolized through glycolysis, the pentose phosphate pathway, or other associated metabolic routes. These pathways generate reducing equivalents, predominantly in the form of NADH and NADPH, which are essential for facilitating chromate reduction89,90. The electrons produced during glucose oxidation are subsequently transferred to a series of intracellular electron carriers, including quinones, flavins, and cytochromes, which serve as intermediates within the bacterial electron transport chain91. Glucose can provide a large number of electrons through glycolysis for the enzymatic reduction of Cr(VI). During glucose fermentation, the formation of volatile fatty acids (VFAs) can also enhance the performance of enzymatic reduction of Cr(VI) to Cr(III)87,92.
In the present study, genes encoding for enzymes responsible for chromate reduction, i.e., chromate reductase (ChrR), azoreductase, and FMN-dependent NADH-azoreductase type1, were detected in the genome of SA1. Such a group of chromate-reducing enzymes has a promising role in the reduction of toxic Cr(VI) to less toxic Cr(III)21. In aerobic conditions, the chromate reductase enzyme transfers an electron from the oxidation of NADH or NADPH to Cr(VI) to generate Cr(V). This intermediate unstable state subsequently takes two electrons from other co-substances to produce less toxic Cr(III)21,93,94. Unlike membrane-bound chromate reductases, soluble reductases are more appropriate for creating biocatalysts for bioremediation applications93. An earlier study also reported that Bacillus subtilis has an azoreductase activity that confers defence against the lethal effects of Cr(VI) and also catalyzes the reduction of Cr(VI)21. In support of this notion, a significant finding of the present study was the presence of chromate reductase genes in the genome of the studied bacterium confirming its Cr(VI) resistance property.
Conclusion
This study represents a pioneering effort to investigate Cr(VI) resistance, reduction potential, and removal capabilities of indigenous bacterial strains from this region. The identification and characterization of potent Cr(VI)-resistant native bacteria capable of efficient chromate reduction is crucial for developing microbial consortia aimed at the bioremediation of Cr-contaminated environments. Bioremediation strategies that leverage indigenous microorganisms offer several advantages, including cost-effectiveness, ease of implementation, and minimal generation of secondary pollution. However, environmental parameters like pH, temperature, nutrient availability, co-contaminants, and biotic interactions significantly influence bacterial survival and metabolic activity. A key determinant of bacterial resistance to heavy metals lies in the presence of specific resistance genes, located either chromosomally or on plasmids. Horizontal gene transfer (HGT) may facilitate the dissemination of metal resistance genes alongside antibiotic resistance determinants, complicating efforts to isolate genes solely responsible for Cr(VI) detoxification. Despite these challenges, the genetic engineering of indigenous bacteria already adapted to complex and variable environmental conditions holds promise as a practical and effective bioremediation approach. Advances in molecular biology and biotechnological tools now enable the extraction of key resistance genes from native strains, paving the way for the development of genetically enhanced bacteria with improved heavy metal detoxification capabilities. The present work provides new insights into Cr(VI) resistance in indigenous Bacillus sp., underscoring their potential for application in the remediation of Cr(VI)-contaminated soils and aquatic systems. Integrative studies involving plant growth-promoting rhizobacteria, whole-genome sequencing, transcriptomics, and untargeted metabolomics are further required to deepen our understanding of chromate resistance mechanisms and the broader ecological functionality of these indigenous bacterial strains.
Data availability
All data related to the manuscript is present within the article itself and the associated supplementary materials.
References
Sharma, B., Dangi, A. K. & Shukla, P. Contemporary enzyme based technologies for bioremediation: A review. J. Environ. Manage. 210, 10–22 (2018).
Verma, S. & Kuila, A. Bioremediation of heavy metals by microbial process. Environ. Technol. Innov. 14, 100369 (2019).
Adhikary, S., Saha, J., Dutta, P. & Pal, A. Bacterial homeostasis and tolerance to potentially toxic metals and metalloids through diverse transporters: Metal-Specific insights. Geomicrobiol J. 41 (5), 496–518 (2024).
Pal, A., Bhattacharjee, S., Saha, J., Sarkar, M. & Mandal, P. Bacterial survival strategies and responses under heavy metal stress: a comprehensive overview. Crit. Rev. Microbiol. 48 (3), 327–355 (2022).
Shahid, M. et al. Chromium speciation, bioavailability, uptake, toxicity and detoxification in soil-plant system: A review. Chemosphere 178, 513–533 (2017).
Sharma, P., Singh, S. P., Parakh, S. K. & Tong, Y. W. Health hazards of hexavalent chromium (Cr (VI)) and its microbial reduction. Bioengineered 13 (3), 4923–4938 (2022).
Vareda, J. P., Valente, A. J. M. & Durães, L. Assessment of heavy metal pollution from anthropogenic activities and remediation strategies: A review. J. Environ. Manage. 246, 101–118 (2019).
El Messaoudi, N. et al. Comprehensive analytical review of heavy metal removal efficiency using agricultural solid waste-based bionanocomposites. Nano-Structures Nano-Objects. 38, 101220 (2024).
Upadhyay, N. et al. Tolerance and reduction of Chromium(VI) by Bacillus sp. MNU16 isolated from contaminated coal mining soil. Front. Plant Sci. 8, 778 (2017).
Gu, R. et al. Chromium metabolism characteristics of coexpression of ChrA and ChrT gene. Ecotoxicol. Environ. Saf. 204, 111060 (2020).
Baldiris, R., Acosta-Tapia, N., Montes, A., Hernández, J. & Vivas-Reyes, R. Reduction of hexavalent chromium and detection of chromate reductase (ChrR) in Stenotrophomonas maltophilia. Molecules 23 (2), 406 (2018).
Elahi, A., Arooj, I., Bukhari, D. A. & Rehman, A. Successive use of microorganisms to remove chromium from wastewater. Appl. Microbiol. Biotechnol. 104, 3729–3743 (2020).
Jobby, R. & Desai, N. Bioremediation of heavy metals. Biodegradation Bioremediat. Environ. Sci. Eng. 8, 201–220 (2017).
Jobby, R., Jha, P., Yadav, A. K. & Desai, N. Biosorption and biotransformation of hexavalent chromium [Cr (VI)]: a comprehensive review. Chemosphere 207, 255–266 (2018).
Dubey, P., Farooqui, A., Patel, A. & Srivastava, P. K. Microbial innovations in chromium remediation: mechanistic insights and diverse applications. World J. Microbiol. Biotechnol. 40 (5), 151 (2024).
Zhu, Y., Yan, J., Xia, L., Zhang, X. & Luo, L. Mechanisms of cr (VI) reduction by Bacillus sp. CRB-1, a novel cr (VI)-reducing bacterium isolated from tannery activated sludge. Ecotoxicol. Environ. Saf. 186, 109792 (2019).
Igiri, B. E. et al. Toxicity and bioremediation of heavy metals contaminated ecosystem from tannery wastewater: a review. J. Toxicol. 2018. (2018).
Mala, J. G. S., Sujatha, D. & Rose, C. Inducible chromate reductase exhibiting extracellular activity in Bacillus methylotrophicus for chromium bioremediation. Microbiol. Res. 170, 235–241 (2015).
Mishra, S. et al. A kinetic modelling approach to explore mechanism of cr (VI) detoxification by a novel strain Pseudochrobactrum saccharolyticum NBRI-CRB 13 using response surface methodology. World J. Microbiol. Biotechnol. 40 (10), 288 (2024).
Ramli, N. N., Othman, A. R., Kurniawan, S. B., Abdullah, S. R. S. & Hasan, H. A. Metabolic pathway of cr (VI) reduction by bacteria: a review. Microbiol. Res. 268, 127288 (2023).
Valenzuela-García, L. I. et al. Novel biochemical properties and physiological role of the flavin mononucleotide oxidoreductase YhdA from Bacillus subtilis. Appl. Environ. Microbiol. 86 (20), e01688–e01620 (2020).
Thatoi, H., Das, S., Mishra, J., Rath, B. P. & Das, N. Bacterial chromate reductase, a potential enzyme for bioremediation of hexavalent chromium: a review. J. Environ. Manage. 146, 383–399 (2014).
Sanjay, M. S., Sudarsanam, D., Raj, G. A. & Baskar, K. Isolation and identification of chromium reducing bacteria from tannery effluent. J. King Saud University-Science. 32 (1), 265–271 (2020).
Tirkey, S. R., Ram, S., Chandrashekhar, P. & Mishra, S. Bioremediation of soils polluted with hexavalent chromium using Bacteria. In: Industrial Wastewater Reuse: Applications, Prospects and Challenges. 249–266. (Springer,2023).
Chen, Y. S., Yanagida, F. & Shinohara, T. Isolation and identification of lactic acid bacteria from soil using an enrichment procedure. Lett. Appl. Microbiol. 40 (3), 195–200 (2005).
Lever, M. A. et al. A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types. Front. Microbiol. 6, 476 (2015).
Strejcek, M., Smrhova, T., Junkova, P. & Uhlik, O. Whole-cell MALDI-TOF MS versus 16S rRNA gene analysis for identification and dereplication of recurrent bacterial isolates. Front. Microbiol. 9, 1294 (2018).
Roy, V. et al. Isolation, characterization, identification, genomics and analyses of bioaccumulation and biosorption potential of two arsenic-resistant bacteria obtained from natural environments. Sci. Rep. 14 (1), 5716 (2024).
McGinnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32 (suppl_2), W20–W25 (2004).
Saha, J., Dey, S. & Pal, A. Whole genome sequencing and comparative genomic analyses of Pseudomonas aeruginosa strain isolated from arable soil reveal novel insights into heavy metal resistance and codon biology. Curr. Genet. 68 (3–4), 481–503 (2022).
Parte, A. C., Sardà Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C. & Göker, M. List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. MicroBiol. 70 (11), 5607–5612 (2020).
Parte, A. C. LPSN–List of prokaryotic names with standing in nomenclature (bacterio. net), 20 years on. Int. J. Syst. Evol. MicroBiol. 68 (6), 1825–1829 (2018).
Sievers, F. & Higgins, D. G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27 (1), 135–145 (2018).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35 (6), 1547–1549 (2018).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591 (2007).
Kepenek, E. S., Severcan, M., Gozen, A. G. & Severcan, F. Discrimination of heavy metal acclimated environmental strains by chemometric analysis of FTIR spectra. Ecotoxicol. Environ. Saf. 202, 110953 (2020).
Faghihzadeh, F., Anaya, N. M., Schifman, L. A. & Oyanedel-Craver, V. Fourier transform infrared spectroscopy to assess molecular-level changes in microorganisms exposed to nanoparticles. Nanatechnol. Environ. Eng. 1 (1), 1–16 (2016).
Afzal, A. M., Rasool, M. H., Waseem, M. & Aslam, B. Assessment of heavy metal tolerance and biosorptive potential of Klebsiella variicola isolated from industrial effluents. AMB Express. 7 (1), 1–9 (2017).
Saha, J., Adhikary, S. & Pal, A. Analyses of the heavy metal resistance pattern and biosorption potential of an Indigenous Bacillus tropicus strain isolated from arable soil. Geomicrobiol J. 39 (10), 891–905 (2022).
Sau, G. B., Chatterjee, S. & Mukherjee, S. K. Chromate reduction by cell-free extract of Bacillus firmus KUCr1. Pol. J. Microbiol. 59 (3), 185 (2010).
Mohamed, M. S., El-Arabi, N. I., El-Hussein, A., El-Maaty, S. A. & Abdelhadi, A. A. Reduction of chromium-VI by chromium-resistant Escherichia coli FACU: a prospective bacterium for bioremediation. Folia Microbiol. 65, 687–696 (2020).
Waterborg, J. H. The Lowry method for protein quantitation. The Protein Protocols Handbook 2009 7–10 .
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic Acids Res. 51 (D1), D418–D427 (2023).
Choudhary, S. & Sar, P. Characterization of a metal resistant Pseudomonas sp. isolated from uranium mine for its potential in heavy metal (Ni2+, Co2+, Cu2+, and Cd2+) sequestration. Bioresour. Technol. 100 (9), 2482–2492 (2009).
Ayangbenro, A. S., Babalola, O. O. & Aremu, O. S. Bioflocculant production and heavy metal sorption by metal resistant bacterial isolates from gold mining soil. Chemosphere 231, 113–120 (2019).
Blum, M. et al. InterPro: the protein sequence classification resource in 2025. Nucleic Acids Res. 53 (D1), D444–D456 (2024).
Fischl, A. S. & Kennedy, E. P. Isolation and properties of acyl carrier protein phosphodiesterase of Escherichia coli. J. Bacteriol. 172 (9), 5445–5449 (1990).
Focardi, S., Pepi, M. & Focardi, S. E. Microbial reduction of hexavalent chromium as a mechanism of detoxification and possible bioremediation applications. Biodegradation-life Sci. 321–347. ( 2013).
Ontañon, O. M., Fernandez, M., Agostini, E. & González, P. S. Identification of the main mechanisms involved in the tolerance and bioremediation of cr (VI) by Bacillus sp. SFC 500-1E. Environ. Sci. Pollut. Res. 25, 16111–16120 (2018).
Suresh, G. et al. Bioremediation of hexavalent chromium-contaminated wastewater by Bacillus thuringiensis and Staphylococcus capitis isolated from tannery sediment. Biomass Convers. Biorefinery. 11, 383–391 (2021).
Wu, M. et al. Bioreduction of hexavalent chromium using a novel strain CRB-7 immobilized on multiple materials. J. Hazard. Mater. 368, 412–420 (2019).
Tan, H. et al. Bioreduction and biosorption of cr (VI) by a novel Bacillus sp. CRB-B1 strain. J. Hazard. Mater. 386, 121628 (2020).
Nicholson, W. L., Munakata, N., Horneck, G., Melosh, H. J. & Setlow, P. Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol. Mol. Biol. Rev. 64 (3), 548–572 (2000).
Pieper, D. H. & Reineke, W. Engineering bacteria for bioremediation. Curr. Opin. Biotechnol. 11 (3), 262–270 (2000).
Varjani, S. J. & Upasani, V. N. A new look on factors affecting microbial degradation of petroleum hydrocarbon pollutants. Int. Biodeterior. Biodegrad. 120, 71–83 (2017).
Rolfe, M. D. et al. Lag phase is a distinct growth phase that prepares bacteria for exponential growth and involves transient metal accumulation. J. Bacteriol. 194 (3), 686–701 (2012).
Dönmez, G. & Aksu, Z. Bioaccumulation of copper (II) and nickel (II) by the non-adapted and adapted growing Candida Sp. Water Res. 35 (6), 1425–1434 (2001).
Adriano, D. C. Nickel 2nd edn (Springer, 2001).
Parameswari, E., Lakshmanan, A. & Thilagavathi, T. Chromate resistance and reduction by bacterial isolates. Aust. J. Basic Appl. Sci. 3 (2), 1363–1368 (2009).
Tripathi, M. & Garg, S. K. Co-remediation of Pentachlorophenol and Cr6 + by free and immobilized cells of native Bacillus cereus isolate: spectrometric characterization of PCP dechlorination products, bioreactor trial and chromate reductase activity. Process Biochem. 48 (3), 496–509 (2013).
Narayani, M. & Shetty, K. V. Chromium-resistant bacteria and their environmental condition for hexavalent chromium removal: a review. Crit. Rev. Environ. Sci. Technol. 43 (9), 955–1009 (2013).
Mabrouk, M. E. Statistical optimization of medium components for chromate reduction by halophilic Streptomyces sp. MS-2. Afr. J. Microbiol. Res. 2, 103–109 (2008).
Faghihzadeh, F., Anaya, N. M., Schifman, L. A. & Oyanedel-Craver, V. Fourier transform infrared spectroscopy to assess molecular-level changes in microorganisms exposed to nanoparticles. Nanatechnol. Environ. Eng. 1, 1–16 (2016).
Zarnowiec, P., Lechowicz, L., Czerwonka, G. & Kaca, W. Fourier transform infrared spectroscopy (FTIR) as a tool for the identification and differentiation of pathogenic bacteria. Curr. Med. Chem. 22 (14), 1710–1718 (2015).
Satapute, P., Paidi, M. K., Kurjogi, M. & Jogaiah, S. Physiological adaptation and spectral annotation of arsenic and cadmium heavy metal-resistant and susceptible strain Pseudomonas taiwanensis. Environ. Pollut. 251, 555–563 (2019).
Nie, M. et al. Selenium-mediated cr(VI) reduction and senps synthesis accelerated Bacillus cereus SES to remediate cr contamination. J. Hazard. Mater. 457, 131713 (2023).
Sun, Y., Jin, J., Li, W., Zhang, S. & Wang, F. Hexavalent chromium removal by a resistant strain Bacillus cereus ZY-2009. Environ. Technol. 44 (13), 1926–1935 (2023).
Cheung, K. & Gu, J-D. Chromate reduction by Bacillus megaterium TKW3 isolated from marine sediments. World J. Microbiol. Biotechnol. 21, 213–219 (2005).
Cheng, G. & Li, X. Bioreduction of chromium (VI) by Bacillus sp. isolated from soils of iron mineral area. Eur. J. Soil Biol. 45 (5–6), 483–487 (2009).
Mangaiyarkarasi, M. M., Vincent, S., Janarthanan, S., Rao, T. S. & Tata, B. Bioreduction of cr (VI) by alkaliphilic Bacillus subtilis and interaction of the membrane groups. Saudi J. Biol. Sci. 18 (2), 157–167 (2011).
Chen, Z. et al. Cr (VI) uptake mechanism of Bacillus cereus. Chemosphere 87 (3), 211–216 (2012).
Fathollahi, A., Khasteganan, N., Coupe, S. J. & Newman, A. P. A meta-analysis of metal biosorption by suspended bacteria from three phyla. Chemosphere 268, 129290 (2021).
Gu, Y. et al. Cr (VI)-bioremediation mechanism of a novel strain Bacillus paramycoides cr6 with the powerful ability to remove cr (VI) from contaminated water. J. Hazard. Mater. 455, 131519 (2023).
Harboul, K., El Aabedy, A., Hammani, K. & El-Karkouri, A. Reduction of hexavalent chromium using Bacillus safensis isolated from an abandoned mine. Environ. Technol. 45 (22), 4495–4511 (2024).
Gu, T. et al. The characteristics of molasses-based reductive removal of cr (VI) from groundwater by Bacillus Sp. J. Environ. Chem. Eng. 10 (6), 108595 (2022).
Huang, Y. et al. Influencing factors and mechanism of cr (VI) reduction by facultative anaerobic Exiguobacterium sp. PY14. Front. Microbiol. 14, 1242410 (2023).
Samuel, J. et al. The differential stress response of adapted chromite mine isolates Bacillus subtilis and Escherichia coli and its impact on bioremediation potential. Biodegradation 24 (6), 829–842 (2013).
Ndeddy Aka, R. J. & Babalola, O. O. Effect of bacterial inoculation of strains of Pseudomonas aeruginosa, Alcaligenes feacalis and Bacillus subtilis on germination, growth and heavy metal (Cd, Cr, and Ni) uptake of Brassica juncea. Int. J. Phytoremed. 18 (2) 200–209. (2016).
Rao, M. A., Scelza, R., Scotti, R. & Gianfreda, L. Role of enzymes in the remediation of polluted environments. J. Soil. Sci. Plant. Nutr. 10 (3), 333–353 (2010).
Cheung, K., Lai, H. & Gu, J-D. Membrane-associated hexavalent chromium reductase of Bacillus megaterium TKW3 with induced expression. J. Microbiol. Biotechnol. (2006).
Bae, W-C. et al. Purification and characterization of NADPH-dependent cr (VI) reductase from Escherichia coli ATCC 33456. J. Microbiol. 43 (1), 21–27 (2005).
Ackerley, D. et al. nd, Chromate-reducing properties of soluble flavoproteins from Pseudomonas putida and Escherichia coli. Appl. Environ. Microbiol. 70(2):873–882. (2004).
Sandana Mala, J. G., Sujatha, D. & Rose, C. Inducible chromate reductase exhibiting extracellular activity in Bacillus methylotrophicus for chromium bioremediation. Microbiol. Res. 170, 235–241 (2015).
Liu, Z., Wu, Y., Lei, C., Liu, P. & Gao, M. Chromate reduction by a chromate-resistant bacterium, Microbacterium Sp. World J. Microbiol. Biotechnol. 28, 1585–1592 (2012).
Satarupa, D. & Paul, A. Hexavalent chromium reduction by aerobic heterotrophic bacteria Indigenous to chromite mine overburden. Brazilian J. Microbiol. 44, 307–315 (2013).
Ziagova, M., Koukkou, A. & Liakopoulou-Kyriakides, M. Optimization of cultural conditions of Arthrobacter sp. Sphe3 for growth-associated chromate (VI) reduction in free and immobilized cell systems. Chemosphere 95, 535–540 (2014).
Rahman, Z. & Thomas, L. Chemical-assisted microbially mediated chromium (Cr)(VI) reduction under the influence of various electron donors, redox mediators, and other additives: an outlook on enhanced cr (VI) removal. Front. Microbiol. 11, 619766 (2021).
Liu, Y-G., Xu, W-H., Zeng, G-M., Li, X. & Gao, H. Cr (VI) reduction by Bacillus sp. isolated from chromium landfill. Process Biochem. 41 (9), 1981–1986 (2006).
Tweeddale, H., Notley-McRobb, L. & Ferenci, T. Effect of slow growth on metabolism of Escherichia coli, as revealed by global metabolite pool (metabolome) analysis. J. Bacteriol. 180 (19), 5109–5116 (1998).
Rabaey, K., Lissens, G., Siciliano, S. D. & Verstraete, W. A microbial fuel cell capable of converting glucose to electricity at high rate and efficiency. Biotechnol. Lett. 25 (18), 1531–1535 (2003).
Methé, B. A. et al. Genome of Geobacter sulfurreducens: metal reduction in subsurface environments. Science 302 (5652), 1967–1969 (2003).
Zheng, X., Yuan, D., Li, Y. & Liu, C. Exploration of the reduction mechanism of cr (VI) in anaerobic hydrogen fermenter. Environ. Pollut. 254, 113042 (2019).
Karim, M. E. et al. Biotransformation of chromium (VI) by Bacillus sp. isolated from chromate contaminated landfill site. Chem. Ecol. 36 (10), 922–937 (2020).
Thatoi, H. & Paul, M. Bioremediation of highly toxic hexavalent chromium by bacterial chromate reductases family: A structural and functional overview. Bioprospecting Enzymes Ind. Healthc. Sustain. Environ. :29–47. (2021).
Acknowledgements
The authors are grateful to DST-SAIF, IIT Bombay, and DST-SAIF, IIT Madras for their sophisticated analytical services and Barcode Biosciences, Bengaluru, for their DNA sequencing service.
Author information
Authors and Affiliations
Contributions
SA: Conceptualization, Methodology, Investigation, Validation, Formal analysis, Resources and Writing - Original Draft and Revisions. BKS and VR: Conceptualization, Investigation, Formal analysis, Visualization and Writing - Original Draft. JS: Investigation, Methodology, Visualization and Writing. AP: Conceptualization, Methodology, Software, Validation, Investigation, Formal analysis, Resources, Writing - Original Draft and Revisions, Supervision and Project administration. All the authors have thoroughly read the final draft and consented to publish it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All the authors have approved the final manuscript and consented to publish the same.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Adhikary, S., Saha, B.K., Roy, V. et al. Deciphering chromate tolerance and reduction ability of an indigenous Bacillus strain isolated from polluted pond sludge for chromium bioremediation. Sci Rep 15, 23323 (2025). https://doi.org/10.1038/s41598-025-07031-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07031-4
