
Abstract
Dengue virus (DENV) poses a critical health threat causing millions of cases every year. The absence of proof-reading activity of the RNA-dependent RNA polymerases (RdRp) leads to significant mutations in its genome. This may result in genetic diversity among the genotypes which contribute to severe complications during the infection. Targeting these mutations may help to understand the viral transmission and identify a potential treatment. An aggregate of 152 dengue positive serum samples were collected from different tertiary care hospitals in Karachi. RNA isolation, cDNA synthesis as well as amplification of the C-prM region was performed, followed by the Sanger sequencing. The resultant sequences of each serotype were aligned with their respective genome sequences and phylogenetic trees based on genotypes were constructed using Maximum Likelihood (ML) method with 1000 bootstraps replicates. The sequences were then converted to the amino acid sequence and genetic variations in comparison to the reference genomes were evaluated. The results showed that there was a higher number of positive cases in males (58%) than in females (42%). The most affected age group was 21–30. The study suggested that the dominant serotype in the population of Karachi was DENV-1 (78.33%), followed by DENV- 2 (15%) and DENV-3 (6.66%). Overall, the acquired data provide the information about genetic variation in the C-prM region of dengue virus in Karachi, Pakistan.
Similar content being viewed by others
Introduction
Dengue virus is classified within the Flaviviridae family1. The genome of the dengue virus comprises 10.7 kb single-stranded, positive sense RNA, which encodes a single polyprotein, that further cleaves into three structural; Capsid (C), Membrane (M), and Envelop (E), and seven non-structural proteins; NS1, NS2A, NS2B, NS3, NS4A, NS4B, and2. Structural proteins are responsible for the formation of viral particles and the non-structural proteins participate in the replication of viral RNA, assembly of viruses, and modulation of host cell responses3,4.
Based on the differences in antigenic determinants, dengue virus is classified into four serotypes, DENV-1, DENV-2, DENV-3, and DENV-4. These serotypes are further divided into genotypes based on envelop gene sequences, DENV1 (1I-V), six in DENV2 (2I-VI), and four in DENV3 (3I, 3II, 3III, 3 V) and DENV4 (4I-IV)5,6,7. Although the genotyping is primarily based on envelop gene, the C-prM region has also been used in several studies for the classification of genotypes8,9,10. A fifth serotype was found in the Sarawak state of Malaysia in October 2013. DENV serotypes 1–4 follow the human cycle, while DENV-5 follows the sylvatic cycle11. The symptoms caused by these serotypes might be similar but phylogenetically they are distinct12. The genotypes within each serotype are distinctly distributed across different geographical locations. Although all serotypes are competent to cause severe infection, some serotypes are more likely to lead to severe cases than others. Several studies have found that the Asian genotype of DENV-2 can cause more severe infection in comparison to the American genotype as the latter efficiently replicates in human cells as well as can efficiently cause infection in new mosquitoes13,14.
In Pakistan, dengue fever is endemic, and it has become a major public health issue. Pakistan faces sporadic dengue outbreaks each year during the monsoon season. The prevalence of dengue infection has greatly increased in Pakistan over the years, burdening the already compromised economic situation of the country15. Pakistan reported its first dengue case in the year 1994 and then the annual outbreaks started in Karachi in November16. Pakistan features several factors that provide optimal conditions for the growth of Aedes mosquito which include rapid urbanization, lack of public health resources, climate, rainfall, insufficient water storage and waste management facilities17,18,19.
In Pakistan, DENV-2 is the most prevalent serotype, followed by DENV-1 and DENV-320. Several factors are responsible for expanding the reach of dengue vectors, such as globalization, international trade, travel, urbanization, changes in climate, and the lack of efficient vector control programs21. Increasing cases of dengue infections are strongly associated with climate change22. According to23, high temperatures increase the risk of dengue infections.
Due to the presence of various alterations in the dengue virus genome, which have been identified as contributing to its enhanced pathogenicity and transmission, preventing and treating dengue infection is becoming challenging. Therefore, the main objective of this study is to evaluate the genetic variations in the genome of all four dengue virus serotypes. The study also aims at finding the most frequent dengue virus serotypes and genotypes present in the population of Karachi.
Methodology
Ethical approval
The samples used in the study were taken after the approval of the ethical committees from the respective laboratories. This study was approved by the Institutional Ethical Committee (IEC) of the International Center for Chemical and Biological Sciences (ICCBS), University of Karachi (Study # ICCBS/IEC/102-HB/2024/Protocol/1.0). Written informed consent was taken from the patients.
Sample and data collection
One hundred and fifty-two (152) dengue NS1 positive serum samples were collected from different hospitals in Karachi between April to August 2022, along with clinical and demographic data with the consent of the infected patients.
RNA extraction and cDNA synthesis
RNA extraction from serum samples was performed using the QIAamp Viral RNA Mini Kit (Qiagen, Germany), according to the manufacturer’s protocol. RNA quantification was carried out using a nanodrop spectrophotometer, followed by first-strand cDNA synthesis with random hexamer primer, using RevertAid First Strand cDNA Synthesis Kit (ThermoFisher Scientific, USA). The concentration of cDNA was quantified using a Qubit dsDNA High Sensitivity kit. The synthesized cDNA was then stored at -80°C for subsequent experimentation24,25.
Polymerase chain reaction (PCR), agarose gel electrophoresis and PCR product purification
The synthesized cDNA was amplified using two consensus primers, D1-forward primer (5’-TCAATATGCTGAAACGCGCGAGAAACCG-3’) and D2-reverse primer (5’-TTGCACCAACAGTCAATGTCTTCAGGTTC-3’), designed by26.
The PCR reaction was run using Absolute Master Mix (MOLEQULE-ON, New Zealand), which contains buffer, dNTPs mix, Taq polymerase, and loading dyes. Initial denaturation was carried out at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 1 min, extension at 72°C for 2 min, and final extension at 72°C for 5 min. The amplified products were run on 2% agarose gel.
The amplified PCR products were purified using Agencourt Ampure XP beads (Beckman Coulter, USA). Each product was combined with 1.8 × volume of magnetic beads, followed by incubation at room temperature for 3–4 min. The tubes were then placed in the magnetic rack for 2 min. Once the beads were attached to the wall of the tube, the supernatant was discarded. The beads were washed with 90 μL of 80% ethanol in the first round. The ethanol was discarded, and the beads were washed with 100 μL of 80% ethanol. The tubes were dried, and the DNA was eluted using an elution buffer27.
Sanger Sequencing
The purified PCR products were quantified using a Qubit fluorometer (ThermoFisher Scientific, USA). The Sanger sequencing of amplified DNA was carried out using Big Dye Terminator v3.1 Cycle Sequencing Kit (ThermoFisher Scientific, UK) on a conventional thermal cycler. The products were sequenced using D1 (forward primer). A total of 5 μL reaction was prepared, consisting of 0.3 μL BDT, 1 μL sequencing buffer, 1 μL forward primer, cDNA (up to 20ng), and water up to 5 μL. The PCR conditions were set as: Initial denaturation at 95°C for 1 min, followed by 35 cycles of denaturation at 95°C for 12 s, annealing temperature at 50°C for 10 s, and extension at 60°C for 4 min and a final extension at 60°C for 4 min. The sequencing PCR products were purified by using the ethanol precipitation method. The ethanol precipitation method was carried out by mixing the sequencing PCR product with a 5 μL volume of 125 mM ethylenediaminetetraacetic acid (EDTA) and 60 μL absolute ethanol. The tubes were centrifuged at 2200 RCF for 15 min. The supernatant was discarded and 100 μL of freshly prepared 80% ethanol was added and the tubes were centrifuged for 5 min at 2200 RCF. The supernatant was discarded, and the tubes were air dried overnight. The dried pellet was mixed with 10 μL formamide and was mixed well, followed by heat shock at 95°C for 5 min, and finally loaded onto SeqStudio Genetic Analyzer (ThermoFisher Scientific, USA)28.
Sequencing data analysis
The sequenced data was filtered using BioEdit. Low-quality and ambiguous sequences were filtered out to ensure accuracy. The resultant cleaned sequences were then analyzed through BLAST (Basic Local Alignment Search Tool) for serotyping by comparing each sequence with the reference sequences available in the NCBI database. For genotyping, the sequences were subjected to Flavivirus Typing Tool.
Phylogenetic analysis
Reference sequences from different countries of all the genotypes belonging to each serotype were obtained from the NCBI Basic Local Alignment Search Tool (BLAST) to align with the sequenced data. A total of twenty DENV-1 sequences, twenty-nine DENV-2 sequences, and twenty-five DENV-3 sequences were retrieved for the phylogenetic tree construction. The selection of sequences was based on the following criteria: Sequences from dengue endemic and epidemic countries were selected to provide comprehensive understanding of global genotype distribution, for cases with identical mutational patterns from the same year or different years, only one representative sequence per country was selected, and prevalent genotype of each serotype in a given country was prioritized. Phylogenetic trees were constructed using the Maximum Likelihood (ML) method for each serotype of dengue virus based on genotypes, using the General Time Reversible (GTR) model, incorporating both Gamma distribution and a portion of invariant sites (G + I), and the reliability of the corresponding branches was evaluated with 1000 bootstraps replicates. Mega11 software was used for the construction of a phylogenetic tree29, and the tree was visualized by the Interactive Tree of Life (iTOL) website. Mutations in the protein sequences were also evaluated by Mega11 software. The sequences from the current study were compared with the reference genomes. These reference genomes were selected from Pakistan, isolated in different years and the available reference genome at the database ViPR (Virus Pathogen Resource).The sequenced data was then submitted to NCBI GenBank.
Statistical analysis
Data entry, analysis, and processing were performed using IBM SPSS Statistics 21 and Excel. Descriptive analysis including the mean and median age of the studied patients was calculated using Excel.
Results
Gender-wise distribution of samples
This study involved the collection of 152 dengue-positive serum samples, out of which 57.9% (88/152) patients were male, and 42.1% (64/152) patients were female (Fig. 1). The age of the infected patients ranged between 7 months to 75 years (mean age = 23.91 ± 13.05, median age = 23). The age group with the highest rate of dengue prevalence was found to be 21 to 30 years. The mean age found in the female participants was 24.40 ± 13.21 and the median age in females was 24, while in male participants, the mean age was 23.56 ± 12.99 and the median age was 22.

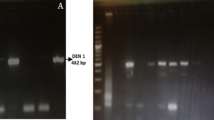
Image of the PCR products subjected to 2% agarose gel electrophoresis, run at 70 V for 70 min, stained with ethidium bromide, and visualized under UV transilluminator. Each resultant PCR product is 511 bp long. Lanes are numbered from left to right; the First lane contains Marker; 100 bp plus ladder used. Lanes 2–19 show PCR products that are 511 bp in size and Lane 20 shows negative control.
PCR amplification using universal primers
The cDNA was amplified using universal primers, D1 (forward) and D2 (reverse). Out of the 152 samples, 100 samples were found to be PCR positive for the C-prM region of the dengue virus as shown in figure-1. The product size was found to be 511 bp.
Distribution of DENV serotypes by sanger sequencing
Samples with high DNA concentration were subjected to Sanger sequencing, revealing a pre-dominance of DENV-1 (78.33%), followed by DENV-2 (15%), and DENV-3.
(6.66%)The visual representation is provided in Fig. 2.

Serotype distribution based on sequencing results among the study population. The graphical chart represents the number of samples identified for different dengue serotypes.
Phylogenetic analysis
DENV-1 analysis
DENV-1 sequences depicted in Fig. 3 show clear branching patterns, with each genotype forming distinct clades. The phylogenetic analysis of the DENV-1 isolates, collected during the 2022 dengue outbreak, revealed that all the isolates clustered within Genotype III. None of the isolates clustered with the reference genomes of Genotypes I (red), IV (orange), and V (purple). The sequences of Genotypes I, IV and V formed distinct clades, indicating clear divergence from the isolates of the present study and highlights genetic diversity of DENV-1 across different geographic locations. The strain OP898557 isolated from Rawalpindi in the year 2022 showed relatedness with the strains (OQ947238, PP800878, OQ947240, OQ947258) from the present study collected from Karachi in the year 2022 (Supp-Table-S1, S2). The 2022 isolates from the current study formed a distinct clade, suggesting either sustained transmission within the region or recent common ancestry and indicating low genetic diversity among circulating strains.

Phylogenetic tree of DENV-1 genotypes, based on partial C-prM region, constructed using the Maximum Likelihood (ML) method with 1000 bootstrap replicates. All the sequences of DENV-1 (47 isolates) from the current study belonged to Genotype III as indicated by green color.
DENV-2 analysis
The comprehensive phylogenetic analysis conducted for DENV-2 isolates, collected during the 2022 dengue outbreak, revealed that all the local strains (green colored) clustered within the Cosmopolitan genotype (blue colored). This monophyletic clade, alongside the other Pakistani strains, suggests continued circulation and evolution of this genotype within the region. The isolates from the current study clustered tightly, showing minimal divergence, indicating strong genetic relatedness and potential local transmission. Furthermore, the earlier isolates from Pakistan within the same cluster support the endemic persistence of the cosmopolitan genotype in the region over the past decade. Alternatively, the reference sequences belonging to Asian I (red), Asian II (orange), American (black), and Asian/American (purple) formed distinct clades, indicating a lack of external introductions (Fig. 4).

Phylogenetic tree of DENV-2 sequences based on C-prM region, constructed using the Maximum Likelihood method, using 1000 bootstrap replicates. The tree shows clear separation of DENV-2 genotypes and highlights persistence of the Cosmopolitan genotype in Pakistan.
DENV-3 analysis
For the construction of DENV-3 phylogenetic tree based on C-prM region, sequences from different geographical regions were included, represented by different color codes as shown in Fig. 5. The analysis revealed that all the 2022 DENV-3 isolates (green colored) clustered with the genotype III reference sequences, but formed their own subgroup, indicating some genetic differences that could be due to recent mutations or local adaptation. The sequences from this study formed a single clade and showed close relatedness with the strains from Myanmar (MW788888, MW788911) (Supp-Table-S1, S2). In contrast, the other genotypes formed distinct clades, representing diverse strains, including those from China, India, Thailand, Myanmar, and South America, suggesting the global spread of DENV-3.

The tree was generated using the Maximum Likelihood (ML) method with 1000 bootstrap replicates, based on the partial C-prM region. The genotypes from the current study are represented in green color.
Mutational analysis
The sequenced samples from the present study showed no insertions or deletions. The amino acid changes observed in the C-prM region of all serotypes are shown in (Supp-Table-3 (A, B, and C)). and the mutations identified and the number of sequenced samples positive for the mutations are depicted in Table-1. In DENV-1, a total of 29 substitutions were observed while 8 substitutions were found in DENV-2 and 7 in DENV-3.
In the genome of DENV-1, multiple mutations were observed, but two unique mutations were found in all 47 sequenced samples: substitution of aspartic acid with glutamic acid at position 176 (D176E) and substitution of valine with isoleucine at position 178 (V178I). The genome of DENV-2 remained largely conserved and displaying only a few mutations, as shown in Table 1. The genome of DENV-3 had five unique mutations found exclusively in the sequenced samples from the current study: the substitution of alanine with valine at position 30, lysine with glutamine at position 31, phenylalanine with valine at position 72, and isoleucine with tyrosine at position 145.
Discussion
Dengue virus has caused numerous outbreaks in Pakistan over the years, infecting thousands of people and contributing significantly to morbidity and mortality. Globally, the incidence of dengue infection has escalated rapidly, placing nearly half of the world’s population at risk30.
The aim of the study was to identify the predominant serotype and genotype of dengue virus in Karachi, circulating in the population during the 2022 outbreak.
Among the studied participants, a higher proportion of dengue infection was observed in male patients (58%) compared to female patients (42%). This disparity may be attributed to outdoor exposure, leading to a greater risk of mosquito bites by the Aedes vector. However, the finding of this study is in contrast to a cross-sectional study conducted in Karachi in the 2021 by31, which reported a significantly higher prevalence of dengue infection in female patients (n = 172, 91%) than in male patients (n = 17, 9%).
Among various age groups, the age group 21 to 30 exhibited the highest incidence of dengue infection. This trend aligns with previous studies32,33,34,35,36, suggesting that this age group may be more susceptible due to environmental and behavioral factors. However, other factors like region, sample size, or study design may also influence this pattern. Further population-based studies are required to confirm this trend on a broader scale.
Serotyping revealed that DENV-1 was the most prevalent serotype in 2022 (78.33%), followed by DENV-2 (15%) and DENV-3 (6.66%). These findings differ from the prior findings in Karachi, where DENV-2 was reported as the dominant serotype15,37,38,39. The recent dominance of DENV-1 may reflect a shift in herd immunity dynamics, where prior widespread immunity to DENV-2 has curtailed its transmission, making the population more susceptible to infection by other dengue serotypes.
The phylogenetic analysis revealed genotype III of DENV-1, cosmopolitan genotype of DENV-2, and genotype III of DENV-3 as the major circulating genotypes in the 2022 outbreak. These observations align with the previous studies, including38, suggesting sustained circulation patterns in the region.
Among the evaluated dengue serotypes, DENV-3 displayed the highest genetic diversity, indicating continuous viral genetic change. This study identified multiple mutations, particularly in DENV-3 and DENV-1, while DENV-2 exhibited relatively fewer changes in the C-prM region. Such mutations may lead to the emergence of new genotypes, possibly modifying transmission efficiency, disease severity, as well as vaccine effectiveness40,41.
The shift from DENV-2 to DENV-1 as the dominant serotype highlights the dynamic nature of the dengue virus and underscores the necessity for continuous genomic surveillance. The identification of these mutations may indicate enhanced pathogenicity and transmission, highlighting the need for robust public health strategies.
Karachi, a densely populated sub-tropical coastal city, presents a conductive environment for dengue transmission due to rapid urbanization combined with several other factors such as hot and humid climate, inadequate sanitation system, and persistent water stagnation, all of which provide ideal breeding grounds for the vector Aedes mosquito. These conditions are escalating public health burden of dengue in Pakistan42.
In conclusion, understanding the genetic variations of circulating DENV is crucial for tracking viral evolution, enhancing preparedness for future outbreaks. The reinforcement of molecular surveillance and vector control programs are key measures to control dengue-related mortalities and morbidities.
Conclusion
This study provides important findings on the distribution of dengue virus serotypes and genotypes during the 2022 outbreak in Karachi, Pakistan. Young adults, particularly those aged between 21 to 30, were the most affected, likely due to increased outdoor exposure and subsequent contact with the Aedes vector. Notably, a higher infection rate was observed in male patients, reflecting gender-based differences in exposure patterns.
Our findings revealed DENV-1 as the dominant serotype (78.33%), highlighting a major shift in serotype prevalence from the historically dominant DENV-2. DENV-2 (15%) and DENV-3 (6.66%) were also detected, indicating the co-circulation of multiple serotypes. Furthermore, the phylogenetic analysis confirmed the presence of Genotype III (DENV-1), Cosmopolitan genotype (DENV-2), and Genotype III (DENV-3), suggesting persistent circulation of established genotypes in Pakistan.
The Sanger sequencing of the C-prM region revealed high conservation across most isolates, however, several mutations were observed, especially in the genome of DENV-3 genome, which could have potential implications on dengue virus pathogenicity and may contribute to emergence of any other genotype in future outbreaks.
These findings underscore the need for continuous genomic surveillance to monitor serotype shift, detect emerging genotypes, and identify mutations with potential clinical relevance. Further studies utilizing whole genome sequencing (WGS) are recommended to gain deeper insights into dengue virus evolution and inform targeted control strategies.
Data availability
The datasets, are available, supporting the conclusions of this article with the accession numbers OQ947232-OQ947237, OQ947239-OQ947270, PP796334-PP796337, PP800870-PP800883, PP796339, and PQ192270 are available in the GenBank [National Center for Biotechnology Information] repository.
References
Frentiu, F. D. Dengue fever: The impact of increasing temperatures and heatwaves. EBioMedicine https://doi.org/10.1016/j.ebiom.2023.104611 (2023).
Poltep, K. et al. Genetic diversity of dengue virus in clinical specimens from Bangkok, Thailand, during 2018–2020: Co-circulation of all four serotypes with multiple genotypes and/or clades. Trop. Med. Infect. Dis. 6(3), 162 (2021).
Kumar, R., Agrawal, T., Khan, N. A., Nakayama, Y. & Medigeshi, G. R. Identification and characterization of the role of c-terminal Src kinase in dengue virus replication. Sci. Rep. 6(1), 1–13 (2016).
Barnard, T. R., Abram, Q. H., Lin, Q. F., Wang, A. B. & Sagan, S. M. Molecular determinants of flavivirus virion assembly. Trends Biochem. Sci. 46(5), 378–390 (2021).
Waman, V. P., Kolekar, P., Ramtirthkar, M. R., Kale, M. M. & Kulkarni-Kale, U. Analysis of genotype diversity and evolution of Dengue virus serotype 2 using complete genomes. PeerJ 4, e2326 (2016).
Wu, T., Wu, Z. & Li, Y. P. Dengue fever and dengue virus in the People’s Republic of China. Rev. Med. Virol. 32(1), e2245 (2022).
Adelino, T. É. R. et al. Field and classroom initiatives for portable sequence-based monitoring of dengue virus in Brazil. Nat. Commun. 12(1), 2296 (2021).
Luo, Z. F. et al. Laboratory and molecular characterization of Dengue Viruses in a 2014 Outbreak in Guangfo Region, Southern China. Jpn. J. Infect. Dis. 70(5), 528–535 (2017).
Islam, A. et al. Circulation of dengue virus serotypes in hyperendemic region of New Delhi, India during 2011–2017. J. Infect. Public Health 13(12), 1912–1919 (2020).
Datu, A. M. et al. Molecular detection and analysis of Dengue virus genetic diversity in North Sulawesi, Indonesia during 2022. Biodiv. J. Biol. Div. https://doi.org/10.13057/biodiv/d240636 (2023).
Mustafa, M. S., Rasotgi, V., Jain, S. & Gupta, V. J. M. J. A. F. I. Discovery of fifth serotype of dengue virus (DENV-5): A new public health dilemma in dengue control. Med. J. Armed Forces India 71(1), 67–70 (2015).
Whitehorn, J. & Simmons, C. P. The pathogenesis of dengue. Vaccine 29(42), 7221–7228 (2011).
Guzman, M. G., Gubler, D. J., Izquierdo, A., Martinez, E. & Halstead, S. B. Dengue infection. Nat. Rev. Dis. Primers. 2(1), 1–25 (2016).
Bhatt, P., Sabeena, S. P., Varma, M. & Arunkumar, G. Current understanding of the pathogenesis of dengue virus infection. Curr. Microbiol. 78(1), 17–32 (2021).
Hanan, F., Ahmad, J., Hadi, S., Ali, I., Fahim, M., and Qayum, A. Analysis of Dengue Virus Genotypes and Further Investigations for Mixed Infections through RT-PCR in Classical Dengue Fever Patients in Pakistan. International Journal of Pathology, 72–78 (2022).
Waheed, S. (n.d.). Dengue fever. Retrieved from https://www.emro.who.int/pak/programmes/dengue-fever.html.
Jahan, F. Dengue fever (DF) in Pakistan. Asia Pac. Fam. Med. 10, 1–4 (2011).
Nabi, G., Ali, M., Khan, S. & Kumar, S. The crisis of water shortage and pollution in Pakistan: Risk to public health, biodiversity, and ecosystem. Environ. Sci. Pollut. Res. 26, 10443–10445 (2019).
Yousaf, M. Z., Siddique, A., Ashfaq, U. A. & Ali, M. Scenario of dengue infection & its control in Pakistan: An up—date and way forward. Asian Pac J Trop Med 11(1), 15–23 (2018).
Ali, L. et al. An overview of dengue viral infection circulating in Pakistan. J. Vector Borne Dis. 59(2), 109–114 (2022).
Huang, Z., Das, A., Qiu, Y. & Tatem, A. J. Web-based GIS: the vector-borne disease airline importation risk (VBD-AIR) tool. Int. J. Health Geogr. 11(1), 1–14 (2012).
Liu-Helmersson, J., Stenlund, H., Wilder-Smith, A. & Rocklöv, J. Vectorial capacity of Aedes aegypti: Effects of temperature and implications for global dengue epidemic potential. PLoS ONE 9(3), e89783 (2014).
Damtew, Y. T. et al. Effects of high temperatures and heatwaves on dengue fever: A systematic review and meta-analysis. EBioMedicine https://doi.org/10.1016/j.ebiom.2023.104582 (2023).
Tazeen, A. et al. Occurrence of co-infection with dengue viruses during 2014 in New Delhi, India. Epidemiol. Infect. 145(1), 67–77 (2017).
Faiz, S. et al. Study of drug resistance-associated genetic mutations, and phylo-genetic analysis of HCV in the Province of Sindh, Pakistan. Sci. Rep. 13(1), 12213 (2023).
Lanciotti, R. S., Calisher, C. H., Gubler, D. J., Chang, G. J. & Vorndam, A. V. Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction. J. Clin. Microbiol. 30(3), 545–551 (1992).
Werner, A., Schäfer, S., Gleußner, N., Nimmerjahn, F. & Winkler, T. H. Determining immunoglobulin-specific B cell receptor repertoire of murine splenocytes by next-generation sequencing. STAR Protoc. 3(2), 101277 (2022).
Fatima, Z. et al. Serotype and genotype analysis of dengue virus by sequencing followed by phylogenetic analysis using samples from three mini outbreaks-2007–2009 in Pakistan. BMC Microbiol. 11, 1–8 (2011).
Isa, I. et al. Genetic diversity of Dengue virus serotypes circulating among Aedes mosquitoes in selected regions of northeastern Nigeria. One Health 13, 100348 (2021).
Qureshi, H. et al. Prevalence of dengue virus in Haripur district, Khyber Pakhtunkhwa, Pakistan. J. Infect. Public Health 16(7), 1131–1136 (2023).
Mushtaq, S. & Abro, M. T. Dengue Cases Presenting to the Emergency Department of a Tertiary Care Hospital in Late 2021: A Cross-Sectional Study in Karachi. Int. J. Pub. Health 69, 1606753 (2024).
Zohra, T. et al. Demographic and clinical features of dengue fever infection in Pakistan: A cross-sectional epidemiological study. Trop. Dis., Trav. Med. Vaccines 10(1), 11 (2024).
Qamash, T. et al. Epidemiological study of dengue fever in district Swabi, Khyber Pakhtunkhwa, Pakistan. Brazilian J. Biol. 81(2), 237–240 (2020).
Mushtaq, S., Khan, M. I. U., Khan, M. T. & Husain, A. Demographic and clinical variables in the dengue epidemic in Punjab, Pakistan. Pak. J. Med. Sci. 39(6), 1742 (2023).
Ali, Z. et al. Prevalence of dengue fever during 2011–2012 in Punjab. J. Anim. Plant Sci. 25(3), 348–354 (2015).
Gupta, E., Dar, L., Kapoor, G. & Broor, S. The changing epidemiology of dengue in Delhi, India. Virol. J. 3, 1–5 (2006).
Khan, E. et al. Co-circulations of two genotypes of dengue virus in 2006 out-break of dengue hemorrhagic fever in Karachi, Pakistan. J. Clin. Virol. 43(2), 176–179 (2008).
Umair, M. et al. Genomic characterization of dengue virus outbreak in 2022 from Pakistan. Vaccines 11(1), 163 (2023).
Umair, M. et al. Serotype diversity of dengue virus reveals the predominance of type 2 in Pakistan during 2019. J. Med. Virol. 92(12), 2900–2902 (2020).
Samune, Y. et al. Genetic regions affecting the replication and pathogenicity of dengue virus type 2. PLoS Negl. Trop. Dis. 18(1), e0011885 (2024).
Díaz, Y. et al. Molecular epidemiology of dengue in Panama: 25 years of circulation. Viruses 11(8), 764 (2019).
Siddiqui, T. R., Ghazal, S., Bibi, S., Ahmed, W. & Sajjad, S. F. Use of the health belief model for the assessment of public knowledge and household preventive practices in Karachi, Pakistan, a dengue-endemic city. PLoS Negl. Trop. Dis. 10(11), e0005129 (2016).
Acknowledgements
We gratefully acknowledge the support provided by Hospitals and diagnostic centers in Karachi for the collection of samples.
Funding
All the experimentations were performed by the institutional recurring grant provided to SF.
Author information
Authors and Affiliations
Contributions
Faaria Tariq-FT-Methodology, Investigation, Data Curation, Bioinformatics Analysis Writing – Original Draft Preparation. Muhammad Irfan-MI-Co-Supervision, Investigation, , Writing – Review and Editing. Saba Farooq- SF-Supervision, Investigations, Formal Analysis, Writing – Review and Editing . Hana’a Iqbal- HI-Validations, Writing –Review and Editing. Atia-tul-Wahab- ATW-Conceptualization, Writing – Review and Editing. Ishtiaq Ahmed- IA-Validations, Writing –Review and Editing. Thomas Iftner- TI-Formal Analysis, Writing –Review and Editing. M. Iqbal Choudhary- MIC-Co-Supervision, Conceptualization, Validation – Review and Editing. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The samples used in the study were taken after the approval of the ethical committees from the respective laboratories. This study was approved by the Institutional Ethical Committee (IEC) of the International Center for Chemical and Biological Sciences (ICCBS), University of Karachi (Study # ICCBS/IEC/102-HB/2024/Protocol/1.0). All methods were performed in accordance with the relevant guidelines and regulations. Written informed consent was taken from the patients.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tariq, F., Irfan, M., Farooq, S. et al. Dynamics and genetic variation of dengue virus serotypes circulating during the 2022 outbreak in Karachi. Sci Rep 15, 22703 (2025). https://doi.org/10.1038/s41598-025-07606-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07606-1
