
Abstract
Mutations in MYH9 and MYH14 are associated with autosomal dominant, progressive sensorineural hearing loss. This study aimed to characterize and compare the clinical and audiological features of patients with MYH9 or MYH14 variants. Thirteen patients with MYH9 or MYH14 mutations were identified through whole-exome or targeted sequencing and underwent audiometric evaluations. Both groups exhibited late-onset, high-frequency, progressive hearing loss. The MYH9 group showed a higher proportion of severe-to-profound cases (41.7%) compared to the MYH14 group (14.3%). One MYH14 case presented with congenital hearing loss linked to a nonsense variant (p.Q25*), expanding the phenotypic spectrum of MYH14-related hearing loss. Several novel variants were identified in both genes, all of which were extremely rare and predicted to be deleterious by in silico analyses. Contrary to previous reports, no syndromic features were observed in patients with MYH9 mutations, although one patient had a marginally elevated mean platelet volume. These findings highlight overlapping auditory phenotypes between MYH9 and MYH14 mutations, with particularly marked variability in the genetic and phenotypic features in MYH9. Early genetic diagnosis can aid in prognosis and support individualized management for patients with progressive hearing loss caused by variants in non-muscle myosin genes.
Similar content being viewed by others
Introduction
Hearing loss is one of the most common sensory disorders, and a significant proportion of cases are attributed to genetic mutations that impair the structure and function of the inner ear1,2. Among the many proteins involved in cochlear integrity and function, members of the myosin superfamily play essential roles in maintaining auditory health. The unconventional myosins, including MYO3A, MYO6, MYO7A, and MYO15A have been well documented for their contribution to hereditary hearing impairment3,4,5. In particular, non-muscle myosins—such as MYH9, MYH10, and MYH14, which constitute non-muscle myosin II isoforms A, B, and C, respectively—are key regulators of cytoskeletal dynamics, influencing cell shape, motility, and adhesion6,7.
The MYH9 gene, located on chromosome 22q12.3, spans 41 exons and is broadly expressed in sensory hair cells and their stereocilia within the cochlea8. Mutations in MYH9 are notably associated with autosomal dominant non-syndromic hearing loss, known as DFNA179. However, mutations in MYH9 are most commonly linked with syndromic manifestations, including sensorineural hearing loss, macrothrombocytopenia, granulocyte inclusions, and nephropathy10,11. In contrast, some patients exhibit only macrothrombocytopenia and sensorineural hearing loss, prompting further investigation into phenotypic variability and underlying mechanisms12,13. On the other hand, MYH14, located on chromosome 19q13.33, also consists of 41 exons and is expressed in the neonatal cochlear sensory epithelium14,15. Unlike MYH9, most MYH14 mutations are associated with non-syndromic hearing loss (DFNA4A), while some variants such as p.R941L have been implicated in broader neurological disorders, including peripheral neuropathy and myopathy16. In addition, MYH10 has been reported to be expressed in the developing cochlea, and knockout mice exhibit malformations in various organs17,18. Although MYH10 plays a critical role in mouse development, no human diseases associated with MYH10 mutations have been reported to date.
Although the precise pathogenic mechanisms by which MYH9 and MYH14 mutations result in auditory dysfunction remain to be fully understood, both genes are believed to impact stereocilia structure and mitochondrial dynamics8,19, critical for auditory signal transduction and hair cell survival. Clinically, both gene mutations tend to produce progressive, high-frequency sensorineural hearing loss with autosomal dominant inheritance. This study seeks to delineate the clinical and audiological phenotypes of patients harboring MYH9 and MYH14 mutations within a large cohort of individuals with hereditary hearing loss. Additionally, we report novel variants discovered through genetic screening and provide a comparative analysis of their associated auditory profiles, contributing to an improved understanding of the genetic basis of adult-onset hearing loss.
Results
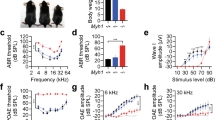
The median age of patients was 58.5 and 54.0 years in the MYH9 and MYH14 mutation groups, respectively. (Table 1) The pure-tone audiogram patterns typically show high-frequency hearing loss and progressive hearing loss. The hearing thresholds of both groups showed no significant differences across all frequencies (Fig. 1a, c,d). However, MYH9 showed a stronger tendency toward severe hearing loss, with 41.7% of cases classified as severe to profound hearing loss, compared to 14.3% in MYH14 (p = 0.190, in Fisher’s exact test). The correlation of hearing function and age was significant in both MYH9 (r = 0.702, p = 0.011) and MYH14 (r = 0.690, p = 0.013; one congenital case was excluded) groups (Fig. 1b). The onset age was mostly after the fifth decade in both mutation groups (50% in MYH9, 57.1% in MYH14).

Audiologic profiles of patients with variants in MYH9 or MYH14. (a) Median pure-tone thresholds at each frequency for patients with MYH9 (blue) and MYH14 (red) variants. Error bars represent interquartile ranges. (b) Correlation between average PTA4 (Y-axis) and age (X-axis). Each dot represents an individual ear, and the lines represent best-fit regression slopes. (c, d) Pure-tone thresholds at each frequency for all enrolled patients are shown. Each dot represents an individual ear. Dots with the same color indicate the same person, and connected lines represent the same ear. X-axis: Pure-tone frequencies (panels A, C, and D); Y-axis: Pure-tone thresholds (dB HL). The dotted line represents 0 dB HL.
Among the 6 affected probands with MYH9 variants, two variants (p.R1730C and p.Q1323H) were known, and three variants (p.Y422C, p.D513Y, and p.S1457F) were novel. The three variants were deleterious in silico and affected an evolutionarily conserved residue across species. Moreover, the variants were observed in only one (p.Y422C) or none (p.D513Y and p.S1457F) of the 1,614,030 alleles in gnomAD v4.1.0. Given that mutations in MYH9 may cause macrothrombocytopenia, platelet count, and mean platelet volume (MPV) were compared, and found to be within the normal range for all evaluated patients (Table 2). However, YUHL410-21, who carries p.R1730C in MYH9, exhibited a high but marginally normal MPV (11.3 fL, normal range: 7.5–11.5 fL) in the absence of overt thrombocytopenia. None of the patients in either mutant group exhibited syndromic features such as nephropathy. In YUHL 107 and 91 families, patients with progressive hearing loss were segregated with the variants in MYH9 (Fig. 2).

Segregation analysis in patients with MYH9 variants. Pedigrees, pure-tone audiograms in affected individuals, and segregation analysis from Sanger sequencing for MYH9 variants are depicted in YUHL 107 and YUH 91 families (3 family members included in each family). In pure-tone audiograms, red and blue colors indicate hearing thresholds at right and left ears, respectively.
Among 7 affected probands with MYH14 variants, six possible variants causing sensorineural hearing loss were identified. (Table 3) Of these, three variants (p.K898Q, p.A1023V, and p.Q25*) were previously reported20,21,22. To strengthen the validity of our findings, we included the same data from patient YUHL77-21 (p.A1023V), which was previously reported by our group. This patient was originally described in the study by Song et al. in 2020 and is reanalyzed here in the context of expanded data22. The other three variants (p.D696N, p.V832I, and p.R879Q) were novel variants, which were classified into variants of unknown significance (VUS) according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines. However, they were predicted as deleterious in silico and the residues were highly conserved across species. In addition, their allele frequencies were rare (minor allele frequency < 0.5E-5), suggesting the likelihood of pathogenic variants for progressive hearing loss.
Discussion
This study provides further evidence that mutations in MYH9 and MYH14 genes contribute to progressive, high-frequency, autosomal dominant hearing loss. Although there is an exceptional case of prelingual, congenital hearing loss caused by a nonsense mutation in MYH14, patients with MYH9 and MYH14 mutations generally exhibit similar auditory profiles. These profiles are commonly characterized by late-onset, gradually progressive hearing impairment, consistent with previous studies. However, a higher percentage of individuals with MYH9 mutations experienced severe-to-profound hearing loss compared to the MYH14 group, suggesting that MYH9 variants may lead to more aggressive auditory deterioration in some patients. Interestingly, MYH9 mutations appeared to be associated with a more severe hearing phenotype. Nearly 42% of MYH9 cases progressed to severe-to-profound hearing loss, compared to just 14% in the MYH14 cohort. Although this difference did not reach statistical significance, likely due to the small sample size, it suggests that MYH9 mutations may, in some cases, result in more aggressive auditory deterioration.
MYH9-associated cases revealed a broader range of onset ages, including early adulthood, further indicating genetic and phenotypic heterogeneity. The observed p.R1730C mutation in two unrelated MYH9 patients may reflect a mutation hotspot or founder effect, though functional studies are necessary to confirm this hypothesis. Syndromic features such as macrothrombocytopenia and nephropathy are commonly reported in MYH9-related disorders, but none of the patients exhibited such manifestations in this cohort. Although one MYH9 patient (YUHL410-21) showed a marginally elevated MPV, it remained within normal limits, which may indicate either an incomplete penetrance or a non-syndromic variant. These findings align with reports of isolated macrothrombocytopenia in specific MYH9 mutations, particularly those located in the non-helical tail domain23. However, there is a chance that other syndromic features including macrothrombocytopenia and nephritis might not have been fully evaluated or yet at the subclinical level in these patients. Therefore, further investigation with long-term follow-up in a larger cohort would warrant the convincing conclusion about the possibility of nonsyndromic hearing loss caused by specific variants in MYH9.
Notably, one MYH14 case (YUHL1167-21) demonstrated congenital hearing loss, a rare phenotype not commonly associated with MYH14, which typically manifests in adulthood. This variant (p.Q25*) was a nonsense mutation, likely contributing to the early onset due to truncated protein expression. MYH14 variants are typically associated with postlingual, progressive hearing loss. However, some studies have reported prelingual, severe-to-profound hearing loss linked to MYH14 variants. For example, Kim et al. identified two MYH14 variants in Korean families associated with this phenotype20. Notably, both probands presented with congenital or prelingual severe hearing loss. One of these, p.D191G in the head domain, was the first missense mutation associated with early-onset hearing loss. The other, p.Q25*, a nonsense mutation in exon 2, likely resulted in a null allele, contributing to the severe auditory phenotype observed. Similarly, Duman et al. reported novel MYH14 variants in families with autosomal dominant sensorineural hearing loss24, while two of the identified variants (p.D191H and p.D191N) affected the same codon in the previous report but showed postlingual and progressive hearing loss in contrast. Their findings further expanded the phenotypic spectrum of MYH14 mutations, indicating that specific variants (though residue sites are same) can lead to prelingual, severe hearing impairment.
Our data include various missense variants located in different domains: the motor domain (two in MYH9 and two in MYH14), the neck region (three in MYH14), and the tail region (four in MYH9 and one in MYH14). Although the mechanism underlying hearing loss is not fully understood, both MYH9 and MYH14 are commonly expressed in sensory hair cells and are believed to play critical roles in maintaining structural integrity8,15. Furthermore, MYH9 is also expressed in supporting structures such as the spiral ligament and Reissner’s membrane, suggesting its potential importance in auditory conduction8,25. Verver et al. additionally reported that mutations in the tail domain are associated with a milder form of hearing loss compared to those in the motor domain26. However, we were unable to confirm this trend in our study, possibly due to the limited number of enrolled patients and the lack of age-matched audiometric data. The domain-specific molecular mechanisms of MYH9 and MYH14 should be further investigated in future studies using animal models.
This study emphasizes the need for extensive genetic testing in adult patients with idiopathic progressive hearing loss. Early identification of MYH9 or MYH14 mutations can provide vital prognostic information, influence surveillance for syndromic features, and guide personalized hearing rehabilitation strategies. Furthermore, the shared auditory phenotype between the two gene groups highlights the potential for overlapping molecular mechanisms, likely involving actin-myosin interactions and mitochondrial function in sensory hair cells.
In conclusion, our findings broaden the phenotypic spectrum associated with MYH9 and MYH14 mutations and emphasize the necessity of variant-level characterization. Future studies involving larger cohorts, longitudinal auditory profiling, and mechanistic investigations will be crucial for developing targeted interventions and enhancing our understanding of genetic hearing loss.
Methods
Patient enrollment
The present study enrolled patients from the cohort for genetic hearing loss, namely Yonsei University Hearing Loss (YUHL) cohort27,28,29,30,31. All patients in the YUHL cohort were audiologically screened to identify those with hearing loss. When family history was noted, available family members also underwent audiological testing. Genetic tests were performed on the affected patients and their family members when a genetic form of hearing loss was suspected. This protocol was followed for all enrolled patients in the study. The inclusion criteria were patients who were genetically confirmed to have pathogenic or likely pathogenic variants in MYH9 or MYH14. Moreover, VUS in MYH9 or MYH14 were additionally considered if allele frequency is exceptionally low, or genotype-phenotype was segregated. As MYH9 or MYH14 may cause late-onset, and progressive hearing loss with both monogenic and polygenic patterns, VUS was also taken into account to investigate the audiological profiles in patients carrying these potential risk alleles. Subsequently, a total of 13 independent patients (6 with MYH9 mutations and 7 with MYH14 mutations) were enrolled in this study. This study was approved by the Institutional Review Board of our hospital (approval no. 4-2015-0659). Written informed consent was obtained from all participants. This study was conducted in accordance with the Declaration of Helsinki.
Evaluation of hearing function
All the enrolled patients and their family members, if available, undertook pure-tone audiometry, as previously reported30,32. Briefly, the pure-tone air (500–4000 Hz) and bone conduction (500–4000 Hz) thresholds were measured using clinical audiometers in a double-walled audio booth. The hearing threshold was calculated as the average threshold at 500, 1000, 2000, 4000, and 8000 Hz (PTA4). The hearing loss was classified into mild (25 ≤ PTA4 < 40 dB HL), moderate (40 ≤ PTA4 < 70 dB HL), and severe-to-profound (PTA4 ≥ 70 dB HL) levels.
Genetic analysis
Genetic testing using next-generation sequencing was performed for individuals and their family members, as described in prior studies. A two-track approach to genetic testing was implemented, utilizing either whole-exome sequencing (WES) or a deafness gene panel, depending on the patient’s insurance coverage and their preference. For panel-based next-generation sequencing, a customized 207-gene deafness panel, as previously described, was employed28,30,33,34. WES was performed using the Agilent SureSelect V5 enrichment capture kit (Agilent Technologies, Santa Clara, CA, USA), following the manufacturer’s protocol for sample preparation. Sequencing was conducted on the MiSeq platform (Illumina, San Diego, CA, USA) with the MiSeq Reagent Kit v2 (300 cycles) for high-throughput sequencing. Sanger sequencing was used for segregation analysis. Variants were identified using the “Basic Variant Caller” function in CLC, requiring a minimum of five reads, 20x coverage, and a frequency of at least 20%. Variants with minor allele frequencies exceeding 0.5% for recessive and 0.05% for dominant hearing loss genes in the dbSNP and gnomAD databases were excluded. Genetic diagnoses were confirmed by a multidisciplinary board of otolaryngologists and clinical geneticists, adhering to the hearing-loss-specific guidelines from the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP), as referenced in the Deafness Variation Database35.
Statistical analysis
All analyses were performed using Prism v8.0 (GraphPad Software, San Diego, CA, USA). Pearson correlation analysis was applied, where r represents the Pearson correlation coefficient. A P value of < 0.05 was considered statistically significant.
Data availability
The datasets generated and analysed during the current study are available in the BioProject repository, accession number PRJNA125667.
References
Gadenstaetter, A. J., Krumpoeck, P. E. & Landegger, L. D. Inner ear gene therapy: an overview from bench to bedside. Mol. Diagn. Ther. 29, 161–181 (2025).
Nicolson, T. Navigating hereditary hearing loss: pathology of the inner ear. Front Cell. Neurosci 15, 660812 (2021).
Friedman, T. B., Belyantseva, I. A. & Frolenkov, G. I. Myosins and hearing. Adv. Exp. Med. Biol. 1239, 317–330 (2020).
Miyoshi, T., Belyantseva, I. A., Sajeevadathan, M. & Friedman, T. B. Pathophysiology of human hearing loss associated with variants in myosins. Front Physiol 15, 1374901 (2024).
Moreland, Z. G. et al. Myosin-based nucleation of actin filaments contributes to stereocilia development critical for hearing. Nat Commun 16(1), 947 (2025).
Chinthalapudi, K. & Heissler, S. M. Structure, regulation, and mechanisms of nonmuscle myosin-2. Cell Mol. Life Sci 81(1), 263 (2024).
Quintanilla, M. A., Hammer, J. A. & Beach, J. R. Non-muscle myosin 2 at a glance. J Cell. Sci 136(5), jcs260890 (2023).
Mhatre, A. N., Li, Y., Atkin, G., Maghnouj, A. & Lalwani, A. K. Expression of Myh9 in the mammalian cochlea: localization within the stereocilia. J. Neurosci. Res. 84, 809–818 (2006).
Lalwani, A. K. et al. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin. Am. J. Hum. Genet. 67, 1121–1128 (2000).
Balduini, C. L., Pecci, A. & Savoia, A. Recent advances in the Understanding and management of related inherited thrombocytopenias. Brit J. Haematol. 154, 161–174 (2011).
Strasser, B. & Haushofer, A. Macrothrombocytopenia and granulopoieitic Döhle body-like inclusions in a MYH9-related disorder. J. Hematop. 17, 235–236 (2024).
Pan, C. et al. A novel MYH9 mutation related to non-syndromic delayed post-lingual sensorineural hearing loss. Eur. Arch. Oto-Rhino-L. 279, 2811–2817 (2022).
Verver, E. et al. R705H mutation of is associated with related disease and not only with non-syndromic deafness DFNA17. Clin. Genet. 88, 85–89 (2015).
Yang, T. et al. Genetic heterogeneity of deafness phenotypes linked to DFNA4. Am. J. Med. Genet. A. 139a, 9–12 (2005).
Donaudy, F. et al. Nonmuscle myosin heavy-chain gene is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am. J. Hum. Genet. 74, 770–776 (2004).
Choi, B. O. et al. A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in. Hum. Mutat. 32, 669–677 (2011).
Ma, X. F., Bao, J. J. & Adelstein, R. S. Loss of cell adhesion causes hydrocephalus in nonmuscle myosin II-B-ablated and mutated mice. Mol. Biol. Cell. 18, 2305–2312 (2007).
Yamamoto, N., Okano, T., Ma, X. F., Adelstein, R. S. & Kelley, M. W. Myosin II regulates extension, growth and patterning in the mammalian cochlear duct. Development 136, 1977–1986 (2009).
Almutawa, W. et al. The R941L mutation in disrupts mitochondrial fission and associates with peripheral neuropathy. Ebiomedicine 45, 379–392 (2019).
Kim, B. J. et al. Discovery of MYH14 as an important and unique deafness gene causing prelingually severe autosomal dominant nonsyndromic hearing loss. J. Gene. Med. 19, e2950 (2017).
Miyagawa, M., Naito, T., Nishio, S., Kamatani, N. & Usami, S. -i. Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PloS One. 8, e71381 (2013).
Song, M. H. et al. Genetic inheritance of late-onset, down-sloping hearing loss and its implications for auditory rehabilitation. Ear Hear. 41, 114–124 (2020).
Pecci, A. et al. MYH 9-Related disease: A novel prognostic model to predict the clinical evolution of the disease based on Genotype–Phenotype correlations. Hum. Mutat. 35, 236–247 (2014).
Duman, D. et al. Identification of novel MYH14 variants in families with autosomal dominant sensorineural hearing loss. Am. J. Med. Genet. A. 194, e63563 (2024).
Lalwani, A. K. et al. A new locus for nonsyndromic hereditary hearing impairment, DFNA17, maps to chromosome 22 and represents a gene for cochleosaccular degeneration. Am. J. Hum. Genet. 64, 318–323 (1999).
Verver, E. J. J. et al. Nonmuscle myosin heavy chain IIA mutation predicts severity and progression of sensorineural hearing loss in patients with related disease. Ear Hear. 37, 112–120 (2016).
Han, J. H. et al. Characterization of vestibular phenotypes in patients with genetic hearing loss. J. Clin. Med. 13, 2001 (2024).
Jung, J. et al. Clinical characteristics and audiological profiles of patients with pathogenic variants of WFS1. J. Clin. Med. 13, 4851 (2024).
Kang, M. et al. Novel variant in CEP250 causes protein mislocalization and leads to nonsyndromic autosomal recessive type of progressive hearing loss. Cells 12, 2328 (2023).
Nam, J. et al. Natural history of auditory function in patients with Alport syndrome: A case series study. J. Clin. Med. 13, 6639 (2024).
Rim, J. H. et al. Differential genetic diagnoses of adult post-lingual hearing loss according to the audiogram pattern and novel candidate gene evaluation. Hum Genet, 141, 915–927 (2022).
Joo, S. Y. et al. Clinical heterogeneity associated with MYO7A variants relies on affected domains. Biomedicines 10, 798 (2022).
Han, J. H. et al. Comprehensive prediction model, including genetic testing, for the outcomes of cochlear implantation. Ear Hear. 44, 223–231 (2023).
Kim, J. A. et al. Systematic genetic assessment of hearing loss using whole-genome sequencing identifies pathogenic variants. Experimental & Mol. Medicine, 57(4), 775–787 (2025).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613 (2018).
Kim, S. J. et al. Genetic association of MYH genes with hereditary hearing loss in Korea. Gene 591, 177–182 (2016).
Acknowledgements
We would like to express our special thanks to the Clinical Research Coordinators, Da Hye Kim and Young Eun Kim, for their invaluable help in collecting patient samples.
Funding
This research was supported by the National Research Foundation of Korea grant funded by the Korea government (MSIT) (RS-2024-00346485 to J.J.) and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number : RS-2024-00439403 to J.J.).
Author information
Authors and Affiliations
Contributions
B.S.H., J.S.Y., G.H.Y., W.D., and J.J. performed experiments, analyzed data and wrote the paper. J.S.H. analyzed the variants identified in this work and revised the manuscript. K.S.H., C.J.Y., W.D., and G.H.Y. collected data. J.J. designed and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bae, S.H., Joo, S.Y., Jang, S.H. et al. The audiological phenotype of patients with a variant in MYH9 and MYH14 genes. Sci Rep 15, 22324 (2025). https://doi.org/10.1038/s41598-025-08801-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-08801-w
This article is cited by
-
Auditory genotype-phenotype correlation of patients with variants in STRC
Scientific Reports (2025)
