
Abstract
Males and females exhibit differences in proteome profiles associated with disease risk. However, sex-dimorphic protein quantitative trait loci (SD-pQTL) and their effects on sex differences in health disorders have not been thoroughly investigated. We conducted a sex-stratified, genome-wide association study on 2,922 proteins using data from 30,272 individuals of Caucasian ancestry from the UK Biobank and compared the estimated effects on protein levels of these variants in the men and women to identify SD-pQTLs. The identified SD-pQTLs were replicated using data from two Japanese cohorts (comprising 2,886 and 1,394 individuals, respectively), as well as from 1,990 Finnish, 630 South Asian, and 662 Black ancestry individuals. Sex-dimorphic pleiotropy and the causal relationship between protein levels and health disorders were assessed using the identified SD-pQTLs. We identified 113 SD-pQTLs associated with 65 proteins. Of the 113 SD-pQTLs, 52 were significant in both sexes, five were not significant in either sex, and 42 and 14 were significant only in males and females, respectively. Variant rs2270416 was significantly associated with the CDH15 protein in both sexes but showed opposite effect direction in men and women. Of the 113 SD-pQTLs identified, a total of 41 were replicated in a meta-analysis encompassing Japanese, South Asian, and Black ancestry individuals. SD-pQTLs for proteins APOE (rs157581) and SNAP25 (rs4420638) exhibited sex-dimorphic associations with dementia, indicating sex dimorphic pleiotropy in both proteins and health disorders. From sex-stratified Mendelian randomization using the SD-pQTLs, proteins NCAM1 and PZP showed significant causal relationship with dementia in males and females, respectively. The present study provides evidence of sex-dimorphic genetic architecture in protein-level regulation, elucidating the proteo-genetic architecture for sex differences in human variation.
Similar content being viewed by others


Introduction
Accounting for sex differences at the molecular level could lead to better personalized disease prediction, diagnosis, and treatment, as well as an improved understanding of the biological mechanisms driving these differences. Mounting evidence suggests differences between males and females in various aspects of health disorders, including the effects of risk factors, prevalence, and disease outcomes1,2,3,4,5,6,7,8,9,10,11,12,13. At the molecular level, sex-differentiated architectures and effects have been investigated at the genomic, transcriptomic, and epigenomic levels, providing insight into sex differences14,15,16,17,18,19,20,21,22,23,24,25.
In recent years, there has been growing evidence of sex differences in protein levels and the differential effects of genetic variants on protein levels26,27,28. For example, a study involving 1,277 European brains indicated differences in protein expression between sexes, as well as genetic variants affecting protein levels differently according to sex28. Another study involving 800 individuals from a Dutch population reported sex-dimorphic genetic regulation of inflammatory proteins, providing broad insights into sex differences in proteo-genetic architecture29. These studies have, however, been limited in terms of the number of traits and proteins studied as well as the number of individuals and ancestries represented.
The objectives of the present study were to identify sex-dimorphic protein quantitative trait loci (SD-pQTLs) and examine their association with sex differences in health disorders. To achieve this, we analyzed 2,922 proteins using data from 30,272 individuals of Caucasian ancestry from the UK Biobank. Next, we replicated the identified SD-pQTLs using five different datasets: (1) 2,886 and (2) 1,394 individuals of Japanese ancestry from the BioBank Japan and the Japan COVID-19 Task Force, respectively, (3) 1,990 individuals of Finnish ancestry from FinnGen, as well as (4) 630 individuals of South Asian ancestry (Indian, Pakistani, and Bangladeshi) and (5) 662 individuals of Black ancestry (African and Caribbean) from the UK Biobank. In addition, we assessed the sex-dimorphic effects of SD-pQTLs on health disorders by conducting sex-stratified GWAS for 30 long-term conditions using 338,568 individuals, distinct from those included in the proteomics analysis.
Results
Sex differences in proteome profiles and regulation
A total of 30,272 individuals were included in the proteome analysis. The mean age ± SD was 57.1 ± 7.9 years. 46.2% (13,974 individuals; mean age ± SD: 57.3 ± 8.1) were males and 53.8% (16,298; 57.0 ± 7.8) were females. Of the 2,922 proteins, 2,249 showed a significant association with sex (false discovery rate (FDR)-corrected p-value, termed q, < 0.05, Supplementary Table 1). To estimate the heritability of proteins in each sex and the genetic correlation of proteins between the sexes, we assessed the effect of variants on blood protein levels within each sex by performing sex-stratified genome-wide association study (GWAS) using the 30,272 individuals. As a result, 1,612 proteins showed significant heritability in both males and females. 194 and 348 proteins showed sex-specific significant heritability in males and females, respectively (q < 0.05, Supplementary Table 1). 1,818 proteins showed a significant genetic correlation differing from zero between males and females (q < 0.05, Supplementary Table 1).
Identification of sex-dimorphic pQTL
After selecting index variants from the sex-stratified GWAS using linkage disequilibrium clumping, 31,753 and 36,979 pQTLs were identified in males and females, respectively (q < 0.05, Supplementary Tables 2 and 3). By comparing the effects of variants between males and females across the genome, we derived 113 index pQTLs that exhibit significantly different effects on the levels of 65 proteins by sex, henceforth termed sex-dimorphic pQTLs, or SD-pQTLs (q < 0.05, Supplementary Table 4). 25 out of the 113 SD-pQTLs were not significant in the sex-combined GWAS, which was conducted on protein levels without sex stratification. 52 out of the 113 SD-pQTLs were significant in both sexes, while 42 and 14 were significant only in males and females, respectively (q < 0.05). The remaining five SD-pQTLs were sex-dimorphic but not significant in either sex. Among the 52 SD-pQTLs exhibiting a significant effect in both sexes, variant rs2270416, associated with CDH15, was a stop-gain variant in CDH15 and the only variant with a sex-antagonistic effect. rs2270416 was masked in the sex-agnostic analysis, as it was not significant in the sex-combined GWAS (sex-dimorphic test: q = 2.07E-27; beta in males: -0.23, q = 4.63E-11; beta in females: 0.26, q = 3.01E-17; sex-combined test q = 0.957).
Replication of the identified SD-pQTL
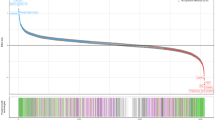
To assess the confidence of the identified SD-pQTLs, we conducted sex-stratified analyses using independent datasets of multiple ancestries. In the BioBank Japan dataset, only 76 out of the 113 SD-pQTLs could be tested because either the proteins were not available or the genetic variants were not polymorphic in BioBank Japan. Among the 76 SD-pQTLs, 12 exhibited significant sex-dimorphic (q-value < 0.05) effects on the protein levels. In the Japan COVID-19 Task Force dataset, 80 out of the 113 SD-pQTLs were testable. Among the 80 SD-pQTLs, three showed significant sex-dimorphic effects. In the FinnGen dataset, 13 of the 111 tested SD-pQTLs showed significant effects. In the UK Biobank South Asian samples, two out of 90 tested SD-pQTLs were significant. In the UK Biobank Black samples, none of the 113 tested SD-pQTLs were significant (Supplementary Table 4). One SD-pQTL (Protein PAEP: variant rs67944) was replicated in BioBank Japan, Japan COVID-19 Task Force, FinnGen, and UK Biobank South Asian samples. Two SD-pQTLs (EDDM3B: rs12890226 and LEFTY2: rs360076) were replicated in BioBank Japan and Japan COVID-19 Task Force. Seven SD-pQTLs (INSL3: rs1044303, KLK3: rs10993994, rs2569747, rs266849, rs266869, PLA2G2A: rs12044628, and PLB1: rs34590437) were replicated only in BioBank Japan, nine (DDR1: rs1264344, EDDM3B: rs4982354, KLK4: rs79486581, NCAM1: rs748631, PAEP: rs10858128, PZP: rs11048434, rs2277413, SUSD4: rs7526539, and TEX101: rs7259375, rs35033974, rs2355990) were replicated only in FinnGen, and one (PZP: rs11615443) was replicated only in UK Biobank South Asian samples. A meta-analysis encompassing all datasets resulted in 22 out of the 113 SD-pQTLs yielding higher confidence than the UK Biobank analysis alone, suggesting that these 22 SD-pQTLs are robust associations (Fig. 1). The sex-dimorphic effect of the variant rs2270416 on CDH15 did not show higher significance in the meta-analysis (p-value = 4.18E-06) than in the UK Biobank alone (p-value = 2.89E-35).

The effects of variants on protein levels in males and females. Each point represents a variant, while lines indicating the 95% confidence interval. The beta values and confidence intervals correspond to the UK Biobank Caucasian ancestry analysis results. Points are highlighted in red when the sex-dimorphic p-value is more significant in the multi-ancestry meta-analysis than in the Caucasian-only UK-Biobank analysis.
SD-pQTLs’ sex-dimorphic effect on health disorders
Genetic variants exhibiting a sex-dimorphic effect on protein levels might also influence health disorders in a sex-dimorphic manner, highlighting the importance of considering sex differences in genetic studies for improving precision medicine and understanding disease mechanisms. To investigate whether the 113 SD-pQTLs in this study also exhibit sex-dimorphic effects on health disorders, we first conducted sex-stratified GWAS on 30 long-term conditions using the 338,568 Caucasian individuals, independent of the samples used in the proteomic GWAS analyses (Supplementary Tables 5 and 6). We then derived results for the 113 SD-pQTLs. As a result, two out of the 113 SD-pQTLs exhibited a significant sex-dimorphic effect on health disorders (p-value < 1.47E-05, Bonferroni corrected p-value accounting for the 113 SD-pQTL and the 30 long-term conditions). SD-pQTLs of the proteins APOE (rs157581, chr19: 45,395,714) and SNAP25 (rs4420638, chr19: 45,422,946) exhibited a sex dimorphic effect on dementia, suggesting potential sex-dimorphic pleiotropy involving these proteins and dementia (Fig. 2). Although it did not reach a significant threshold after correction for multiple testing, the variant rs2270416, which showed a sex dimorphic effect on CDH15, also exhibited a suggested sex dimorphic effect on depression (p-value = 3.29E-02). Sex-specific survival analysis between APOE and SNAP25 measured plasma protein levels and prospective dementia showed a significant association for APOE both in males (hazard ratio (HR) = 0.68, p-value = 3.89E-10) and females (HR = 0.67, p-value = 5.64E-13), but with non-significant sex-dimorphic effect (p-value = 0.94). SNAP25 showed a significant association with prospective dementia only in females (HR = 1.24, p-value = 3.17E-04) but not in males (HR = 1.01, p-value = 0.838).

Sex dimorphic pleiotropy between proteins and health disorders. Scatterplot of variants’ sex-dimorphic effect on proteins and health disorders. Each point represents a variant, with lines indicating the 95% confidence interval. The X-axis and Y-axis represents the difference in the effect on protein and health disorders, between males and females, respectively, with a higher value indicating a higher effect in females. The label on each point describes the associated protein and health disorder for that variant.
We also questioned whether proteins can, in a causal way, differentially influence disease risk depending on sex. This can shed light on possible sex-dimorphic disease mechanisms, that are potentially responsible for sex disparities in drug effectiveness and safety. One such example is the higher risk of adverse drug reactions observed in females, which has been attributed to sex-agnostic drug prescription practices30. Mendelian randomisation (MR) can infer causal relationships between an exposure, such as protein level, and a disease outcome using genetic variants as instrumental variables. That is, genetic variants that are associated with the outcome only through their effect on the exposure. Rather than mere correlation, where no cause-and-effect relationship is implied, MR can estimate causality between exposure and outcome. This is achieved by leveraging the random allocation of genetic variants at birth, thus minimising confounding and reverse causation from outcome to exposure. In light of this, we investigated possible sex-dimorphic causal effects of proteins on health disorders by conducting sex-stratified MR analyses using the identified SD-pQTLs. Here, we conducted MR using seven proteins that have more than one valid SD-pQTL in both sexes and 30 long-term conditions (Fig. 3 and Supplementary Table 7). In contrast to our previous analysis of sex-dimorphic effects on disorders (Fig. 2), sex-stratified MR has the advantages to assess causality and whether or not SD-pQTL effects on disorders are driven by pleiotropy. We identified four protein-disorder pairs where a causal relationship was observed in only one sex. For male-specific relationships, SUSD4-inflammatory bowel disease (Inverse variance weighted (IVW) fixed effects meta-analysis MR estimate q-value in males = 0.038, in females = 1.00) and NCAM1-dementia (q-value in males = 0.045, q-value in females = 1.00) pairs were identified. For female-specific relationships, TSPAN8-asthma (beta in females = 0.84, q-value in females = 2.52E-04) and PZP-dementia (beta in females = 0.96, q-value in females = 0.040) pairs were identified. We did not observe protein-disorder pairs where the male and female-specific causal estimates differed significantly (t-test between male and female estimates q-value > 0.05). No protein-disorder pair showed evidence of heterogeneity or pleiotropy (Supplementary Tables 8 and 9). For the four protein-disorder pairs identified through sex-specific MR, we further fit sex-specific survival analyses. We did not find a significant association between protein levels and prospective disease onset of the tested health disorders in either males or females. Cox Proportional Hazard models were fit for SUSD4-inflammatory bowel disease (protein HR p-value in males = 0.52, in females = 0.35), NCAM1-dementia (HR p-value in males = 0.76, HR p-value in females = 0.61), TSPAN8-asthma (HR p-value in males = 0.37, HR p-value in females = 0.66), PZP-dementia (HR p-value in males = 0.87, HR p-value in females = 0.41). In summary, we identified protein-disorder pairs causally affecting health disorder risk in a male-only or female-only way. No protein-disorder pairs showed significant difference in their sex-stratified causal effects here. These findings may guide future research on potential sex-specific role of those proteins in disease pathogenesis.

Sex specific protein-disorder causal relationships from SD-pQTLs. Scatterplot of sex-stratified MR for protein-disorder pairs. Each point represents a pair, with lines indicating the 95% confidence interval. The X-axis and Y-axis represent causal estimates in males and females, respectively. Blue-coloured pairs indicate male-specific relationships, while red coloured pairs indicate female-specific relationships. The label on each point provides the protein and health disorder of the pair.
Discussion
Our study investigated sex differences in the proteo-genetic architecture of blood proteins across ancestries, providing a comprehensive assessment at a depth and breadth not previously achieved. The findings provide insight into different relationships between genetic variants, protein levels, and health disorders depending on sex. The replication of the identified SD-pQTLs across independent datasets of various ancestries strengthens the validity of our findings.
Our analysis revealed SD-pQTLs that exhibited various differential effects on protein levels between males and females, such as sex-specific or sex-antagonistic effects, as reported in previous sex-stratified GWAS conducted on various human phenotypes16. This suggests that sex differences at the molecular level have complex background constituted of various mechanism that cannot be explained solely by sex-specific or sex-antagonistic pathways. One notable finding from this study is variant rs2270416, which is significantly associated in both sexes, yet exhibits opposite effect directions with CDH15 plasma protein level. This is the only such case in this study, although sex-specific or dimorphic effect of CDH15 have not been clearly suggested to date. It may be implied that this antagonistic association is specific to the individuals in the UK Biobank, considering the lower significance in the meta-analysis or a potential false positive. Hence, further investigation would be required on the SD-pQTLs identified in this study to assess whether the sex-dimorphic effect of each variant is shared or differs across ancestries.
We also found that some SD-pQTLs have been identified as significant pQTLs from sex-agnostic GWAS. This implies that effect size estimates from sex-agnostic GWAS may be inaccurate, as they can represent an average between significantly different effects in males and females. Furthermore, significant associations in sex-agnostics GWAS may reflect strong effects in only one sex, which would go undetected without sex-stratified analysis. These findings highlight the importance of performing sex-stratified GWAS in uncovering sex-dimorphic genetic effects that may be missed in sex-agnostic studies.
In addition, our study provides insights into the sex-dimorphic effects of SD-pQTLs on diseases risk. For instance, we identified SD-pQTLs associated with dementia, exhibiting sex dimorphic effect on disease risk. Blood levels of APOE and SNAP25 have been reported to be associated with dementia and suggested as biomarkers for dementia31,32,33,34. Further investigation with the identified SD-pQTL of APOE and SNAP25 in this study could provide insight into the pathophysiology of dementia and help improve its prediction, diagnosis, and treatment. Additionally, our Mendelian randomization analysis using SD-pQTLs revealed sex-specific causal relationships between proteins and health disorders. Specifically, the NCAM1 and PZP proteins showed sex-specific causal relationships with dementia in males and females, respectively. This finding may provide insight into sex-specific pathophysiology of dementia through further investigation of their biological pathways. Blood levels of these two proteins have previously been reported to be associated with dementia35,36,37. Here, we provide additional genetically informed support for these associations.
PZP is a protein initially described as a major pregnancy-associated protein, showing elevated levels during pregnancy and higher expression levels in females than in males38,39. It has been reported that only females showed elevated serum PZP levels prior to the onset of Alzheimer’s disease, in line with the female-specific causal relationship between PZP and dementia observed in the present study37. Plasma NCAM1 has also been found to mediate sex-related neurodegeneration differences in cognitively normal adults, potentially suggesting that NCAM1 exerts sex-different neuropathological effects at a pre-dementia stage as well40. A previous study of temporal lobe proteomes reported a significant upregulation of NCAM1 only in the brains of male dementia patients41. Although protein levels can vary between brain and blood tissues, the male-specific association between brain NCAM1 levels and dementia reported in the previous study of temporal lobe proteomes and the male-specific causal relationship between serum NCAM1 and dementia in the present study could provide illuminating insight into the sex-dimorphic pathophysiology of dementia. Additionally, NCAM1 has been implicated in the mechanism of action of antidepressants, particularly in response to duloxetine treatment for depression42,43. Studies of pooled data from seven randomised clinical trials found no significant sex differences in duloxetine’s efficacy, safety or tolerability in treating major depressive disorder44,45. However, these duloxetine studies did not evaluate response based on drug dosage or plasma concentrations, which could mask dose-dependent sex differences. Further study on a large cohort, accompanied by experimental analysis, would be necessary to dissect the detailed role of these proteins in dementia. Taken together, these findings highlight the potential role of sex-dimorphic genetic regulation in disease pathogenesis and its implication for sex-specific precision medicine in prediction, diagnosis, and treatment.
However, the results of this study should be interpreted with caution, as we utilized only a limited number of blood proteins currently available, which is just a subset of human proteome. The SD-pQTLs and their associations with health disorders also require careful consideration, as the analysis was limited to Caucasian individuals from the UK Biobank, leveraging its large sample size. Additionally, disease prevalence differs between males and females, as observed in our dataset. Furthermore, the MR investigation of sex-dimorphic causal relationship should also be interpreted with caution, as this study relied solely on SD-pQTLs. Further studies using additional valid genetic instruments for each disease are needed to better dissect sex-dimorphic effects in causal inference. Another limitation of our MR analysis is that it was based on individuals of Caucasian ancestry from the UK Biobank, as SD-pQTLs from other ancestries would have limited power in an MR framework. Restricting MR analyses to ancestrally homogenous samples reduces the risk of population stratification that can lead to violation of the independence and exclusion restriction MR assumptions46. However, transferring MR results across ancestries is challenging due to differences in LD patterns and allele frequencies46, although methods to facilitate trans-ancestry MR have been proposed47. It should also be noted that aside from population stratification, MR analyses can be biased by assortative mating, dynastic events (the direct effect of one’s parents on a phenotype), or selection bias, such as participation bias in the UK Biobank. Such processes can confound the relationship between SD-pQTLs and disease outcomes, potentially violating the MR independence assumption48. Finally, out MR analysis does not account for potential time-varying effects of proteins on disease risk, which may be important when considering the timing of intervention46. Additionally, the limited statistical support in the survival analysis maybe due to a small sample size, specifically the low number of incident dementia events following the date the blood sample was collected for the protein level measurement (288 males and 302 females). Notably, the survival analyses of PZP and NCAM1 on dementia showed the same direction of effect as in the MR, suggesting the possibility of insufficient sample size. Therefore, larger studies with longer follow-up are needed to clarify the sex-dimorphic associations between protein levels and disease incidence observed in this study.
Conclusions
Our study provides comprehensive findings, resources, and insights into sex differences in the proteo-genetic architecture of proteins and their relationships with disease susceptibility. Further research is needed to investigate the underlying mechanisms of sex-dimorphic protein regulation, their links to diseases, and their potential for therapeutic applications.
Methods
Data sources
The UK Biobank is a prospective research resource of population-based cohort study that include comprehensive phenotype and genotype data from approximately 500,000 participants recruited in 2006–2010 residing in England, Scotland, and Wales (www.ukbiobank.ac.uk). This open-access resource was established to support investigations into the factors influencing various health outcomes49. We utilized genotyped data and recently released proteome data from the UK Biobank26. To ensure homogeneity, we limited analyses to unrelated individuals of Caucasian genetic ancestry (UK Biobank Data-Field 22006) with less than 10% missing genotypes and those with matched recorded sex (Data-Field 31) and genetically determined sex (Data-Field 22001). We used this matched sex information to define sex in subsequent analyses. The unrelated participants were identified and extracted using the KING software with following options: --unrelated –degree 2 (version 2.28)50. Autosomal and X-chromosomal genotypes of the selected individuals were filtered using PLINK software (version 1.90b) with the following options; --geno 0.01, --hwe 1e-15, --maf 0.01, and mind 0.1, retaining 539,158 variants51. These filtered variants were used in the subsequent analyses. To ensure homogeneity in proteome analysis, we extracted proteome data of randomly selected baseline participants from protein batches 0–6, which are highly representative of the UK Biobank overall26. Following these selections, the final dataset for proteome analysis consisted of 13,974 males and 16,298 females, totalling 30,272 individuals. The remaining individuals without proteome data, 156,581 males and 181,987 females, totalling 338,568 individuals, were retained, and utilized in subsequent sex-stratified analyses on health disorders.
The BioBank Japan (https://biobankjp.org/en/) project (first cohort) is a hospital-based cohort study that recruited approximately 200,000 patients with at least one of the 47 complex diseases between 2003 and 2007 across 66 hospitals in Japan52. Proteomic profiling was performed on unrelated individuals of East-Asian ancestry from two previous studies with whole genome sequencing datasets, using the Olink Explore 3072 panel following the manufacturer’s protocol53,54. Proteomic data processing and data quality control were conducted according to Olink protocols. The rank-based inverse normal transformation was applied to protein level measurements before association tests for males and females, respectively. Sex-stratified pQTL summary statistics of serum protein levels in the BioBank Japan project were obtained by meta-analysing results for each sex derived from each study separately using REGENIE v3.2.9 (adjusted for age, age2, proteomic profiling batch, and the first 10 genetic principal components) and METAL (fixed-effect inverse variance weighted)55,56. In total, 2,886 participants (including 2,151 males and 735 females) were included in the sex-stratified pQTL analyses in BioBank Japan.
The Japan COVID-19 Task Force (JCTF) was established in early 2020 as a nationwide multicentre consortium to overcome the COVID-19 pandemic (https://www.covid19-taskforce.jp/en/home/). Plasma protein expression was measured using the Olink Explore 3072 platform. We bridge-normalized the Normalized Protein eXpression (NPX) values using the OlinkAnalyze R package with 16 intersecting samples as bridging samples. Samples with QC warning flags were removed. Genotyping was performed using Infinium Asian Screening Array (Illumina, CA, USA), and stringent sample and variant level quality control (QC) filters were applied (sample QC: sample call rate < 0.98, samples of estimated non East Asian ancestry based on PCA with HapMap project samples, variant QC: variant call rate < 0.99, minor allele count < 5, p-value for Hardy-Weinberg equilibrium < 1e-10, and with more than 5% allele frequency difference when compared with the representative reference panels of Japanese ancestry57. We performed genome-wide genotype imputation, by using SHAPEIT4 software version 4.2.1 for haplotype phasing and Minimac4 software version 1.0.1 for genotype imputation58,59. For imputation, we used our in-house and Japanese-specific reference panel composed of N = 4,561 whole-genome sequence (WGS) data from multiple studies (e.g., N = 1,939 from the BBJ study and N = 141 WGS from the previous study)60,61. The final dataset consisted of 995 males and 399 females, totalling 1,394 individuals. The genomic coordinates of the variants are based on the genome build GRCh37 throughout this study.
FinnGen (https://www.finngen.fi/en) is a public-private research project, combining genome and digital healthcare data on ~ 500,000 Finns that launched in 2017. The nation-wide research project is a pre-competitive partnership of Finnish biobanks and their background organisations (universities and university hospitals) and international pharmaceutical industry partners and Finnish biobank cooperative (FINBB). FinnGen aims to provide novel medically and therapeutically relevant insight into human diseases and is described in detail in their flagship paper62. FinnGen partners are listed in full here: https://www.finngen.fi/en/partners.
Investigating sex different genetics in blood proteome
To investigate the effect of sex on proteins, multivariable regression was conducted accounting for age, age2, body mass index, UK Biobank Centre, UK Biobank genetic array, time between blood sampling and measurement, and the first 20 genetic principal components. To investigate genetic effects on blood proteins by sex, we conducted sex-stratified GWAS on protein levels using the proteome data from the 30,272 selected Caucasian individuals and compared variants’ effect on protein between males and females. For the sex-stratified GWAS, REGENIE software (version 3.2.2) was utilized with sex-stratified protein level normalization using --apply-rint option55. Covariates considered in the GWAS included age, age2, batch, UK Biobank Centre, UK Biobank genetic array, time between blood sampling and measurement, and the first 20 genetic principal components. Sex, Sex*age, and sex*age2 were added in covariates for sex-combined GWAS. Protein GLIPR1 was excluded from the analysis due to exceptionally high percent of failing QC (99.40%) during the protein measurement26.
We estimated variant-based heritability as the variance explained by the genetic variants’ effect using the LD-score regression63. To estimate sex difference in heritability, we applied t-statistics utilized in a previous sex stratified study16. We estimated genetic correlation of each protein between males and females using the High-Definition Likelihood method64. To account for multiple testing, we adjusted the p-values using the Benjamini-Hochberg method for each analysis65.
Identification and replication of sex-dimorphic pQTL
To identify variants with different effects between males and females, we compared the effects of each variant across the genome for each protein using two-tailed Student’s t-test, applied in previous sex-stratified GWAS comparison studies16,66,67. To select index variants in each significant locus, we utilized PLINK software with following options: --clump, --clump-p1 0.00000005, --clump-p2 0.001, --clump-r2 0.2, --clump-kb 10,000, considering sex-different p-value as the variant’s significance. Clumping was also applied to GWAS results from each sex as well51. To account for multiple testing, we adjusted the sex different p-values using the Benjamini-Hochberg method, and index variants with false discovery rate < 0.05 were considered significant and presented as the identified SD-pQTLs in this study65. The functional consequences of variants were annotated using Variant Effect Predictor and Ensembl GRCh37 release 11268.
To ensure the confidence of the identified SD-pQTLs, we replicated the identified SD-pQTLs in each independent dataset comprised of 2,886 individuals (2,151 males and 735 females) of Japanese ancestry from the BioBank Japan, 1,394 individuals (995 males and 399 females) of Japanese ancestry from the Japan COVID-19 Task Force, 1,990 (958 males and 1,032 females) of Finnish ancestry from FinnGen, as well as 630 individuals (336 males and 294 females) of South Asian (Indian, Pakistani, and Bangladeshi) and 662 individuals (290 males and 372 females) of Black ancestries (Caribbean and African) in the UK Biobank based on the self-reported ancestry (Data-Field 21000), using the protocols from the identification step in this study. For FinnGen, the 1,990 individuals had plasma proteomics measured using the Olink Explore 3072 panel. Quality control of the proteomics data was carried out in accordance with Olink recommendations by the core FinnGen analysis team. GWAS of plasma protein levels were carried out separately by sex using REGENIE [Ref]. NPX values were rank-based inverse normal transformed (--apply-rint) within sex. Age, batch, genotyping array and genetic PCs (1–5) were included as covariates. A detailed description of FinnGen genotype data, its quality control and pre-processing has been described previously62. FinnGen data freeze 12 was used for this analysis and sex-specific pQTL results were downloaded from the FinnGen sandbox under approved proposal F_2024_056. The replicated results were combined using a random-effect meta-analysis, conducted with the metafor R package and set to “REML”, to obtain a single estimate69. Variants were considered replicated SD-pQTLs if the p-value from the meta-analysis was more significant than that from the UK Biobank analysis alone, indicating a consistent trend in sex-different effect of the variant across independent datasets of multiple ancestries.
Sex dimorphic effect of SD-pQTL on health disorders
To investigate SD-pQTLs’ sex dimorphic effect on health disorder, we firstly conducted sex-stratified GWAS on 30 predefined long-term conditions (Supplementary Table 5) using the 338,568 individuals retained during sample selection70. We then derived results for the 113 SD-pQTLs. For the sex-stratified GWAS, we used the REGENIE software with the following covariates: age, age2, UK Biobank Centre, UK Biobank genetic array, and the first 20 genetic principal components55. Sex-different effects on health disorders were derived using the method applied in SD-pQTL analysis in this study. SD-pQTLs were considered to have sex-different pleiotropy with health disorders if the SD-pQTL exhibited p-value for sex dimorphic effect on health disorder lower than 1.47E-05, which 0.05 divided by the number of long-term conditions and the number of SD-pQTLs, accounting for multiple tests.
To investigate sex-dimorphic causal relationships between plasma protein levels and the 30 predefined health disorders, we employed a two-sample mendelian randomisation (MR) approach separately for males and females71. MR analyses are based on the use of genetic variants as instrumental variables (IV). IVs are variables associated with an exposure but not with the outcome of interest through any other pathway. Three assumptions are required for MR to be valid: (1) IVs are significantly associated with the exposure (the relevance assumption); (2) there are no confounders of the IVs and the outcome (the independence assumption); and (3) IVs do not affect the outcome other than through the exposure (the exclusion restriction assumption)71.
We used the SD-pQTLs identified in this study as instrumental variables. For each MR analysis between protein and health disorder by sex, SD-pQTLs that were significantly associated with each protein (FDR < 0.05) and were not significantly associated with each health disorder (p-value ≥ 5E-8) were selected. SD-pQTLs were used regardless of their genomic position relative to their associated protein’s gene location, i.e. both cis- and trans-pQTLs. Only proteins with more than one SD-pQTLs after filtering were considered for the MR analysis. This was done in order to allow the use of sensitivity analyses testing for horizontal pleiotropy and heterogeneity46.
MR was conducted using the TwoSampleMR R package (version 0.5.11)72. The causal estimates were initially derived using the inverse-variance weighed (IVW) fixed effects meta-analysis method, accompanied by weighted median (WM) and MR-Egger methods73,74,75. WM and MR-Egger analyses were conducted to the proteins with more than 2 valid SD-pQTLs. Sensitivity analyses to test potential horizontal pleiotropy of the causal relationships and heterogeneity of the instrumental variables were conducted using the MR-Egger intercept test and the Cochran’s Q statistic test, respectively75,76,77. Sex dimorphic causal relationships were identified using a two-tailed Student’s t-test and considered as sex-dimorphic if FDR for the sex dimorphic effect is below 0.05. Causal relationships with FDR < 0.05 for either the pleiotropy or heterogeneity tests were excluded.
For the protein-disorder pairs that were significant in the sex-stratified MR analysis, we further examined whether UKB protein levels could predict incident health disorder survival in a sex-dimorphic way. For this purpose, we employed Cox Proportional Hazards models separately for each sex, using baseline protein levels as predictors and time to first incident diagnosis of the respective health disorder as outcome. Survival times were derived based on up to 16 years of follow-up health records. Models were adjusted for age at protein measurement, as it is a confounding factor for many age-related health disorders. Individuals with a prevalent health disorder diagnosis before, or up to 1 year after protein measurement were excluded. The “survival” R package (version 3.7-0) was used to fit the Cox Proportional Hazards models78. All methods have been carried out in accordance with relevant guidelines and regulations.
Data availability
Data of the UK Biobank project in our study is publicly available upon request and approval. Details of procedures for accessing the UK Biobank data can be found here: https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access. The GWAS summary statistics from the UK Biobank dataset and analysis scripts used in this study are publicly available from the University of Edinburgh repository: https://doi.org/10.7488/ds/7917. Whole genome sequence data of BioBank Japan project in this study is available at NBDC database (https://humandbs.dbcls.jp/hum0014-v33). Proteome data and clinical information is available upon request and approval. Details of procedures for accessing Biobank Japan data can be found here: https://biobankjp.org/en/researchers/1974#gsc.tab=0. The summary statistics of pQTL in JCTF will be available upon publication.
Abbreviations
- pQTL:
-
Protein quantitative trait loci
- SD-pQTL:
-
Sex-dimorphic protein quantitative trait loci
- GWAS:
-
Genome-wide association study
- MR:
-
Mendelian Randomization
- APOE:
-
apolipoprotein E
- NCAM1:
-
Neural cell adhesion molecule 1
- PZP:
-
Pregnancy Zone Protein
- SNAP25:
-
Synaptosome Associated Protein 25
References
Mauvais-Jarvis, F. et al. Sex and gender: modifiers of health, disease, and medicine. Lancet 396 (10250), 565–582 (2020).
Heron, M. P. Deaths: leading causes for 2018 (2021).
Agur, K., McLean, G., Hunt, K., Guthrie, B. & Mercer, S. W. How does sex influence multimorbidity? Secondary analysis of a large nationally representative dataset. Int. J. Environ. Res. Public Health. 13 (4), 391 (2016).
Millett, E. R., Peters, S. A. & Woodward, M. Sex differences in risk factors for myocardial infarction: cohort study of UK Biobank participants. bmj 2018, 363 (2018).
Zhao, J. V., Luo, S. & Schooling, C. M. Sex-specific Mendelian randomization study of genetically predicted insulin and cardiovascular events in the UK biobank. Commun. Biol. 2 (1), 332 (2019).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2018. Cancer J. Clin. 68 (1), 7–30 (2018).
Her, A-Y. et al. Sex-specific difference of in-hospital mortality from COVID-19 in South Korea. PLoS One. 17 (1), e0262861 (2022).
Kun, L. et al. Sex-related outcomes of successful drug-coated balloon treatment in de Novo coronary artery disease. Yonsei Med. J. 62 (11), 981 (2021).
de Ritter, R. et al. Sex differences in the risk of vascular disease associated with diabetes. Biol. Sex. Differ. 11, 1–11 (2020).
Ohkuma, T., Komorita, Y., Peters, S. A. & Woodward, M. Diabetes as a risk factor for heart failure in women and men: a systematic review and meta-analysis of 47 cohorts including 12 million individuals. Diabetologia 62, 1550–1560 (2019).
Cordonnier, C. et al. Group wifsie: stroke in women—from evidence to inequalities. Nat. Rev. Neurol. 13 (9), 521–532 (2017).
Bushnell, C. et al. Guidelines for the prevention of stroke in women: a statement for healthcare professionals from the American heart association/american stroke association. Stroke 45 (5), 1545–1588 (2014).
Bhak, Y. et al. Polygenic risk score validation using Korean genomes of 265 early-onset acute myocardial infarction patients and 636 healthy controls. PloS One. 16 (2), e0246538 (2021).
Ober, C., Loisel, D. A. & Gilad, Y. Sex-specific genetic architecture of human disease. Nat. Rev. Genet. 9 (12), 911–922 (2008).
Weiss, L. A., Pan, L., Abney, M. & Ober, C. The sex-specific genetic architecture of quantitative traits in humans. Nat. Genet. 38 (2), 218–222 (2006).
Bernabeu, E. et al. Sex differences in genetic architecture in the UK biobank. Nat. Genet. 53 (9), 1283–1289 (2021).
Flynn, E. et al. Sex-specific genetic effects across biomarkers. Eur. J. Hum. Genet. 29 (1), 154–163 (2021).
Zhu, C. et al. Amplification is the primary mode of gene-by-sex interaction in complex human traits. Cell Genom. 3, 5 (2023).
Lindén, M. et al. Sex influences eQTL effects of SLE and sjögren’s syndrome-associated genetic polymorphisms. Biology Sex. Differences. 8, 1–12 (2017).
Lopes-Ramos, C. M. et al. Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Reports 31, 12 (2020).
Oliva, M. et al. The impact of sex on gene expression across human tissues. Science 369 (6509), eaba3066 (2020).
Yao, C. et al. Sex-and age-interacting eQTLs in human complex diseases. Hum. Mol. Genet. 23 (7), 1947–1956 (2014).
Casazza, W. et al. Sex-dependent placental methylation quantitative trait loci provide insight into the prenatal origins of childhood onset traits and conditions. Iscience 27, 2 (2024).
Zhang, L. et al. Sex-specific DNA methylation differences in alzheimer’s disease pathology. Acta Neuropathol. Commun. 9 (1), 77 (2021).
McCartney, D. L. et al. An epigenome-wide association study of sex-specific chronological ageing. Genome Med. 12, 1–11 (2020).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK biobank. Nature 622 (7982), 329–338 (2023).
de Bakker, M. et al. Sex-based differences in cardiovascular proteomic profiles and their associations with adverse outcomes in patients with chronic heart failure. Biol. Sex. Differ. 14 (1), 29 (2023).
Wingo, A. P. et al. Sex differences in brain protein expression and disease. Nat. Med. 29 (9), 2224–2232 (2023).
Boahen, C. K. et al. Sex-biased genetic regulation of inflammatory proteins in the Dutch population. BMC Genom. 25 (1), 154 (2024).
Franconi, F., Brunelleschi, S., Steardo, L. & Cuomo, V. Gender differences in drug responses. Pharmacol. Res. 55 (2), 81–95 (2007).
Rasmussen, K. L., Tybjærg-Hansen, A., Nordestgaard, B. G. & Frikke‐Schmidt, R. Plasma levels of Apolipoprotein E and risk of dementia in the general population. Ann. Neurol. 77 (2), 301–311 (2015).
Wolters, F. J., Koudstaal, P. J., Hofman, A., van Duijn, C. M. & Ikram, M. A. Serum Apolipoprotein E is associated with long-term risk of alzheimer’s disease: the Rotterdam study. Neurosci. Lett. 617, 139–142 (2016).
Ribaldi, F. et al. Plasma SNAP-25 as a potential alzheimer’s disease biomarker. Alzheimer’s Dement. 19, e076863 (2023).
Agliardi, C. et al. SNAP-25 in serum is carried by exosomes of neuronal origin and is a potential biomarker of alzheimer’s disease. Mol. Neurobiol. 56 (8), 5792–5798 (2019).
Nijholt, D. A. et al. Pregnancy zone protein is increased in the alzheimer’s disease brain and associates with senile plaques. J. Alzheimers Dis. 46 (1), 227–238 (2015).
Gudmundsdottir, V. et al. Serum proteomics reveals APOE dependent and independent protein signatures in alzheimer’s disease (2024).
IJsselstijn, L. et al. Serum levels of pregnancy zone protein are elevated in presymptomatic alzheimer’s disease. J. Proteome Res. 10 (11), 4902–4910 (2011).
Shao, J. et al. Serum Exosomal pregnancy zone protein as a promising biomarker in inflammatory bowel disease. Cell. Mol. Biol. Lett. 26 (1), 36 (2021).
Folkersen, J. et al. Circulating levels of pregnancy zone protein: normal range and the influence of age and gender. Clin. Chim. Acta. 110 (2–3), 139–145 (1981).
Shi, L. et al. Identification of plasma proteins relating to brain neurodegeneration and vascular pathology in cognitively normal individuals. Alzheimer’s Dementia: Diagnosis Assess. Dis. Monit. 13 (1), e12240 (2021).
Gallart-Palau, X. et al. Gender differences in white matter pathology and mitochondrial dysfunction in alzheimer’s disease with cerebrovascular disease. Mol. Brain. 9, 1–15 (2016).
Wędzony, K., Chocyk, A. & Maćkowiak, M. Potential roles of NCAM/PSA-NCAM proteins in depression and the mechanism of action of antidepressant drugs. Pharmacol. Rep. 65 (6), 1471–1478 (2013).
Maciukiewicz, M. et al. Genome-wide association studies of placebo and Duloxetine response in major depressive disorder. Pharmacogenomics J. 18 (3), 406–412 (2018).
Kornstein, S. G., Wohlreich, M. M., Mallinckrodt, C. H., Watkin, J. G. & Stewart, D. E. Duloxetine efficacy for major depressive disorder in male vs. female patients: data from 7 randomized, double-blind, placebo-controlled trials. J. Clin. Psychiatry. 67 (5), 761–770 (2006).
Stewart, D. E., Wohlreich, M. M., Mallinckrodt, C. H., Watkin, J. G. & Kornstein, S. G. Duloxetine in the treatment of major depressive disorder: comparisons of safety and tolerability in male and female patients. J. Affect. Disord. 94 (1–3), 183–189 (2006).
Richmond, R. C. & Smith, G. D. Mendelian randomization: concepts and scope. Cold Spring Harbor Perspect. Med. 12 (1), a040501 (2022).
Hou, L., Wu, S., Yuan, Z., Xue, F. & Li, H. TEMR: Trans-ethnic Mendelian randomization method using large-scale GWAS summary datasets. Am. J. Hum. Genet. 112 (1), 28–43 (2025).
Sanderson, E. et al. Mendelian randomization. Nat. Reviews Methods Primers. 2 (1), 6 (2022).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12 (3), e1001779 (2015).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26 (22), 2867–2873 (2010).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575 (2007).
Nagai, A. et al. Overview of the biobank Japan project: study design and profile. J. Epidemiol. 27 (Supplement_III), S2–S8 (2017).
Koyama, S. et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 52 (11), 1169–1177 (2020).
Liu, X. et al. Decoding triancestral origins, archaic introgression, and natural selection in the Japanese population by whole-genome sequencing. Sci. Adv. 10 (16), eadi8419 (2024).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53 (7), 1097–1103 (2021).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26 (17), 2190–2191 (2010).
Consortium, G. P. An integrated map of genetic variation from 1,092 human genomes. Nature 491 (7422), 56 (2012).
Delaneau, O., Zagury, J-F., Robinson, M. R., Marchini, J. L. & Dermitzakis, E. T. Accurate, scalable and integrative haplotype Estimation. Nat. Commun. 10 (1), 5436 (2019).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48 (10), 1284–1287 (2016).
Okada, Y. et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 9 (1), 1631 (2018).
Sonehara, K. et al. Genetic architecture of MicroRNA expression and its link to complex diseases in the Japanese population. Hum. Mol. Genet. 31 (11), 1806–1820 (2022).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613 (7944), 508–518 (2023).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47 (3), 291–295 (2015).
Ning, Z., Pawitan, Y. & Shen, X. High-definition likelihood inference of genetic correlations across human complex traits. Nat. Genet. 52 (8), 859–864 (2020).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc.: Ser. B (Methodol.). 57 (1), 289–300 (1995).
Winkler, T. W. et al. The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLoS Genet. 11 (10), e1005378 (2015).
Randall, J. C. et al. Sex-stratified genome-wide association studies including 270,000 individuals show sexual dimorphism in genetic loci for anthropometric traits. PLoS Genet. 9 (6), e1003500 (2013).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 1–14 (2016).
Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48 (2010).
Barnett, K. et al. Epidemiology of Multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet 380 (9836), 37–43 (2012).
Hartwig, F. P., Davies, N. M., Hemani, G. & Davey Smith, G. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique 1717–1726 (Oxford University Press, 2016).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, e34408 (2018).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37 (7), 658–665 (2013).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent Estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40 (4), 304–314 (2016).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect Estimation and bias detection through Egger regression. Int. J. Epidemiol. 44 (2), 512–525 (2015).
Bowden, J. et al. Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. Int. J. Epidemiol. 48 (3), 728–742 (2019).
Greco, M. F. D., Minelli, C., Sheehan, N. A. & Thompson, J. R. Detecting Pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat. Med. 34 (21), 2926–2940 (2015).
Therneau, T. M., Grambsch, P. M., Therneau, T. M. & Grambsch, P. M. The Cox Model (Springer, 2000).
Acknowledgements
This work used the Edinburgh Compute and Data Facility (ECDF) (http://www.ecdf.ed.ac.uk/). This research has been conducted using the UK Biobank Resource project 44986. This work uses data provided by patients and collected by the NHS as part of their care and support. This research has been conducted using sample and data from Biobank Japan project. The Japan COVID-19 Task Force is detailed in Supplementary Table 10. We want to acknowledge the participants and investigators of the FinnGen study. The FinnGen project is funded by two grants from Business Finland (HUS 4685/31/2016 and UH 4386/31/2016) and the following industry partners: AbbVie Inc., AstraZeneca UK Ltd, Biogen MA Inc., Bristol Myers Squibb (and Celgene Corporation & Celgene International II Sàrl), Genentech Inc., Merck Sharp & Dohme LCC, Pfizer Inc., GlaxoSmithKline Intellectual Property Development Ltd., Sanofi US Services Inc., Maze Therapeutics Inc., Janssen Biotech Inc, Novartis AG, and Boehringer Ingelheim International GmbH. Following biobanks are acknowledged for delivering biobank samples to FinnGen: Auria Biobank (www.auria.fi/biopankki), THL Biobank (www.thl.fi/biobank), Helsinki Biobank (www.helsinginbiopankki.fi), Biobank Borealis of Northern Finland (https://www.ppshp.fi/Tutkimus-ja-opetus/Biopankki/Pages/Biobank-Borealis-briefly-in-English.aspx), Finnish Clinical Biobank Tampere (www.tays.fi/en-US/Research_and_development/Finnish_Clinical_Biobank_Tampere), Biobank of Eastern Finland (www.ita-suomenbiopankki.fi/en), Central Finland Biobank (www.ksshp.fi/fi-FI/Potilaalle/Biopankki), Finnish Red Cross Blood Service Biobank (www.veripalvelu.fi/verenluovutus/biopankkitoiminta), Terveystalo Biobank (www.terveystalo.com/fi/Yritystietoa/Terveystalo-Biopankki/Biopankki/) and Arctic Biobank (https://www.oulu.fi/en/university/faculties-and-units/faculty-medicine/northern-finland-birth-cohorts-and-arctic-biobank). All Finnish Biobanks are members of BBMRI.fi infrastructure (https://www.bbmri-eric.eu/national-nodes/finland/). Finnish Biobank Cooperative -FINBB (https://finbb.fi/) is the coordinator of BBMRI-ERIC operations in Finland. The Finnish biobank data can be accessed through the Fingenious® services (https://site.fingenious.fi/en/) managed by FINBB. For the purpose of open access, the author has applied a CC-BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Funding
This project was funded by the National Institute for Health Research (NIHR) Artificial Intelligence and Multimorbidity: Clustering in Individuals, Space and Clinical Context (AIM-CISC) grant NIHR202639. BioBank Japan project was supported by AMED under Grant Number JP24tm0624002 and JP243fa627011. This study was also supported by AMED (JP23kk0305022, JP22ek0410075, JP23km0405211, JP23km0405217, JP23ek0109594, JP23ek0410113, JP223fa627002, JP223fa627010, JP233fa627011, JP23zf0127008, JP23tm0524002, JP22fk0108510, JP21fk0108553, JP21fk0108431, JP20fk0108415, JP20fk0108452, JP23tm0524008), JST CREST (JPMJCR20H2), JST FOREST (JPMJFR225Y), JST PRESTO (JPMJPR21R). The super-computing resource was provided by the Human Genome Center (the Univ. of Tokyo). JST Moonshot R&D (JPMJMS2021, JPMJMS2024), MHLW (20CA2054), JSPS KAKENHI (22H00476, 22J30004, 23K14233, 23K27608), the Nakajima Foundation, the Uehara Memorial Foundation, Takeda Science Foundation, the Mitsubishi Foundation, and Bioinformatics Initiative of Osaka University Graduate School of Medicine, Institute for Open and Transdisciplinary Research Initiatives, Center for Infectious Disease Education and Research (CiDER), and Center for Advanced Modality and DDS (CAMaD), Osaka University. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: YB, VR, EM, AM, and AT; Data Analysis: YB, VR, YH, and TN; Data collection: Yoji S, TM, KM, A Kanai, Yutaka S, Yoshiya O, YK, HN, RS, A Kimura, RK, SO, SM, SI, TK, KF, and Yukinori O; Manuscript writing: YB, VR, AM, and AT; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participants
The UK Biobank project was approved by the National Research Ethics Service Committee North West-Haydock (REC reference: 11/NW/0382). Participants provided written informed consent to participate in the UK Biobank. An electronic signed consent was obtained from the participants. This research was conducted using the UK Biobank Resource under project 44986. This work uses data provided by patients and collected by the NHS as part of their care and support. BioBank Japan project was approved by the Institutional Review Board of the University of Tokyo (2023-77-0118 and 2022-61-0119). Informed consent was obtained from all participants. The JCTF was approved by the ethical committees of Keio University School of Medicine, Osaka University Graduate School of Medicine, and affiliated institutes. Informed consent was obtained from all participants. Study subjects in FinnGen provided informed consent for biobank research, based on the Finnish Biobank Act. Alternatively, separate research cohorts, collected prior the Finnish Biobank Act came into effect (in September 2013) and start of FinnGen (August 2017), were collected based on study-specific consents and later transferred to the Finnish biobanks after approval by Fimea (Finnish Medicines Agency), the National Supervisory Authority for Welfare and Health. Recruitment protocols followed the biobank protocols approved by Fimea. The Coordinating Ethics Committee of the Hospital District of Helsinki and Uusimaa (HUS) statement number for the FinnGen study is Nr HUS/990/2017. The FinnGen study is approved by Finnish Institute for Health and Welfare (permit numbers: THL/2031/6.02.00/2017, THL/1101/5.05.00/2017, THL/341/6.02.00/2018, THL/2222/6.02.00/2018, THL/283/6.02.00/2019, THL/1721/5.05.00/2019 and THL/1524/5.05.00/2020), Digital and population data service agency (permit numbers: VRK43431/2017-3, VRK/6909/2018-3, VRK/4415/2019-3), the Social Insurance Institution (permit numbers: KELA 58/522/2017, KELA 131/522/2018, KELA 70/522/2019, KELA 98/522/2019, KELA 134/522/2019, KELA 138/522/2019, KELA 2/522/2020, KELA 16/522/2020), Findata permit numbers THL/2364/14.02/2020, THL/4055/14.06.00/2020, THL/3433/14.06.00/2020, THL/4432/14.06/2020, THL/5189/14.06/2020, THL/5894/14.06.00/2020, THL/6619/14.06.00/2020, THL/209/14.06.00/2021, THL/688/14.06.00/2021, THL/1284/14.06.00/2021, THL/1965/14.06.00/2021, THL/5546/14.02.00/2020, THL/2658/14.06.00/2021, THL/4235/14.06.00/2021, Statistics Finland (permit numbers: TK-53-1041-17 and TK/143/07.03.00/2020 (earlier TK-53-90-20) TK/1735/07.03.00/2021, TK/3112/07.03.00/2021) and Finnish Registry for Kidney Diseases permission/extract from the meeting minutes on 4th July 2019. The Biobank Access Decisions for FinnGen samples and data utilized in FinnGen Data Freeze 12 include: THL Biobank BB2017_55, BB2017_111, BB2018_19, BB_2018_34, BB_2018_67, BB2018_71, BB2019_7, BB2019_8, BB2019_26, BB2020_1, BB2021_65, Finnish Red Cross Blood Service Biobank 7.12.2017, Helsinki Biobank HUS/359/2017, HUS/248/2020, HUS/430/2021 § 28, § 29, HUS/150/2022 § 12, § 13, § 14, § 15, § 16, § 17, § 18, § 23, § 58, § 59, HUS/128/2023 § 18, Auria Biobank AB17-5154 and amendment #1 (August 17 2020) and amendments BB_2021 − 0140, BB_2021 − 0156 (August 26 2021, Feb 2 2022), BB_2021 − 0169, BB_2021 − 0179, BB_2021 − 0161, AB20-5926 and amendment #1 (April 23 2020) and it´s modifications (Sep 22 2021), BB_2022 − 0262, BB_2022 − 0256, Biobank Borealis of Northern Finland_2017_1013, 2021_5010, 2021_5010 Amendment, 2021_5018, 2021_5018 Amendment, 2021_5015, 2021_5015 Amendment, 2021_5015 Amendment_2, 2021_5023, 2021_5023 Amendment, 2021_5023 Amendment_2, 2021_5017, 2021_5017 Amendment, 2022_6001, 2022_6001 Amendment, 2022_6006 Amendment, 2022_6006 Amendment, 2022_6006 Amendment_2, BB22-0067, 2022_0262, 2022_0262 Amendment, Biobank of Eastern Finland 1186/2018 and amendment 22§/2020, 53§/2021, 13§/2022, 14§/2022, 15§/2022, 27§/2022, 28§/2022, 29§/2022, 33§/2022, 35§/2022, 36§/2022, 37§/2022, 39§/2022, 7§/2023, 32§/2023, 33§/2023, 34§/2023, 35§/2023, 36§/2023, 37§/2023, 38§/2023, 39§/2023, 40§/2023, 41§/2023, Finnish Clinical Biobank Tampere MH0004 and amendments (21.02.2020 & 06.10.2020), BB2021-0140 8§/2021, 9§/2021, § 9/2022, § 10/2022, § 12/2022, 13§/2022, § 20/2022, § 21/2022, § 22/2022, § 23/2022, 28§/2022, 29§/2022, 30§/2022, 31§/2022, 32§/2022, 38§/2022, 40§/2022, 42§/2022, 1§/2023, Central Finland Biobank 1-2017, BB_2021 − 0161, BB_2021 − 0169, BB_2021 − 0179, BB_2021 − 0170, BB_2022 − 0256, BB_2022 − 0262, BB22-0067, Decision allowing to continue data processing until 31st Aug 2024 for projects: BB_2021 − 0179, BB22-0067,BB_2022 − 0262, BB_2021 − 0170, BB_2021 − 0164, BB_2021 − 0161, and BB_2021 − 0169, and Terveystalo Biobank STB 2018001 and amendment 25th Aug 2020, Finnish Hematological Registry and Clinical Biobank decision 18th June 2021, Arctic biobank P0844: ARC_2021_1001.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhak, Y., Raptis, V., He, Y. et al. Identification and replication of sex-dimorphic protein quantitative trait loci across multiple ancestries and their associations with diseases. Sci Rep 15, 31721 (2025). https://doi.org/10.1038/s41598-025-10031-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10031-z
