
Abstract
Ischemic stroke (IS) affects 11 million people annually, posing substantial clinical and economic burdens. Current therapies remain limited by time sensitivity and variable efficacy, necessitating novel biomarkers. We developed a multi-omics framework to investigate senescence-associated gene regulation in IS. After normalizing GSE22255 and GSE58294 datasets, we systematically identified aging-related differentially expressed genes (DEGs). Functional annotation via Gene Set Enrichment Analysis (GSEA) and machine learning-driven Weighted Gene Co-expression Network Analysis (WGCNA) identified core genetic signatures validated in the GSE16561 cohort. Key regulators underwent single-gene profiling, immune microenvironment evaluation, and transcriptional network analysis. Reverse transcription-quantitative PCR (RT-qPCR) confirmed bioinformatics findings. We identified 132 senescence-related DEGs, with PTGS2 emerging as a key biomarker. Pathway analyses revealed significant NF-κB, HIF-1, and TGF-β signaling pathway activation. CIBERSORT-based immune profiling showed altered lymphocyte/macrophage ratios in IS patients. Drug-gene interaction analysis identified nine therapeutic compounds, including etodolac targeting NFE2L2 and PTGS2. A strong positive correlation (r = 0.72, p < 0.001) between NFE2L2 and PTGS2 expression was observed. This study establishes senescence-related genes as promising biomarkers and therapeutic targets for IS, particularly through NFE2L2-PTGS2 interactions, providing a foundation for developing immunomodulatory and targeted therapies.
Similar content being viewed by others


Introduction
Ischemic stroke is a major public health concern, characterized by high incidence and disability rates, which poses significant challenges to patients and healthcare systems worldwide1,2. Globally, approximately 11 million individuals suffer from ischemic stroke annually, leading to substantial healthcare costs during the recovery phase1. Current treatment modalities include acute interventions and rehabilitation therapies, yet these approaches are limited by time windows for effective application, variable efficacy, and potential side effects3. As a result, there is a pressing need to explore new biomarkers and therapeutic targets that could enhance diagnosis and treatment outcomes for ischemic stroke patients.
Existing research has established a strong correlation between aging and ischemic stroke, with age-related factors significantly influencing the disease’s pathophysiology4,5,6. Numerous studies have highlighted the involvement of aging-associated genes in the mechanisms underlying ischemic stroke, suggesting that these genes may play critical roles in various biological processes, including inflammation and cellular senescence7,8. Furthermore, recent advancements in molecular biology have underscored the importance of these genes in shaping the disease’s clinical manifestations and outcomes9,10,11. However, despite this progress, there remains a gap in understanding the specific roles of senescence related gene expression profiles in ischemic stroke and how these may serve as potential biomarkers or therapeutic targets.
To address this research gap, our study employs a multi-omics approach, integrating diverse datasets to analyze gene expression patterns and their relationships with clinical features. This methodology includes differential expression analysis, functional enrichment analysis, weighted gene co-expression network analysis (WGCNA), and machine learning techniques. By leveraging these approaches, we aim to provide a comprehensive assessment of potential biomarkers associated with ischemic stroke, with a particular focus on senescence related genes. This integrative analysis not only enhances the understanding of the molecular underpinnings of ischemic stroke but also offers insights into potential therapeutic avenues.
The primary objective of this research is to identify key senescence related genes that are associated with ischemic stroke and to elucidate their potential mechanisms of action and clinical significance. By integrating findings from various omics layers, we hope to uncover novel therapeutic targets that may contribute to improved patient outcomes. This approach is anticipated to shift the current paradigm in ischemic stroke management by highlighting the importance of senescence related biological processes and their implications for treatment strategies.
In conclusion, this study seeks to bridge the existing knowledge gaps regarding the role of senescence related genes in ischemic stroke by employing a robust multi-omics methodology. The insights gained from this research may pave the way for the development of innovative biomarkers and targeted therapies, ultimately improving the diagnosis and treatment of ischemic stroke patients. By addressing the critical relationship between aging and ischemic stroke, this research has the potential to significantly impact clinical practice and enhance patient care in this challenging field.
Materials and methods
Datasets acquisition and preprocessing
In this study, RNA expression profiles and clinical information from the stroke datasets GSE16561, GSE22255, and GSE58294 were retrieved from the Gene Expression Omnibus (GEO) database by the keywords “ischemic stroke”, all the datasets have more than 40 samples. GSE16561 (Platform: Illumina HumanRef-8 v3.0 expression beadchip) comprises 63 samples (39 ischemic stroke patients, 24 controls).GSE22255 (Platform: Affymetrix Human Genome U133 Plus 2.0 Array) includes 40 samples (20 ischemic stroke patients, 20 controls).GSE58294 (Platform: Affymetrix Human Genome U133A Array) encompasses 92 samples (69 ischemic stroke patients of the cardioembolic subtype, 23 controls). During integration of GSE22255 and GSE58294 (both Affymetrix platforms), batch effects were mitigated using the removeBatchEffect function from the R package limma after initial merging via inSilicoMerging.
Differential expression analysis
Following data preprocessing of the two cerebral ischemia-related datasets (GSE22255 and GSE58294), the limma package was employed to delineate differentially expressed genes (DEGs) between ischemic stroke specimens and normal controls. The threshold for defining differential expression was set as a false discovery rate (FDR) < 0.05 combined with absolute log2-transformed fold change values (|log2FC|) exceeding 0.5, which yielded 1,039 significant DEGs. To graphically represent the transcriptional variations, a volcano plot and hierarchical clustering heatmap were subsequently constructed employing the ggplot2 and pheatmap packages, respectively, within the R environment for statistical computing. These visualizations systematically illustrated the distinct patterns in transcriptional profiling data between pathological and control cohorts.
Acquisition of SDEGs
The senescence related gene set utilized in our study was sourced from the Molecular Signatures Database (MSigDB; https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). We conducted a search within the MSigDB database using the key word ‘senescence’ and identified 43 gene sets comprising a total of 2075 genes. Additionally, we acquired 866 senescence-associated DEGs (SDEGs) from the CellAge database (https://genomics.senescence.info/cells/), which were derived from gene manipulation experiments across various human cell types. After eliminating duplicate entries from the two databases, we obtained a final set of 2765 senescence related genes. Through computational intersection of SDEGs with genome-wide differential expression profiles, 132 core molecular signatures were systematically identified, thereby establishing their pivotal roles in cellular senescence mechanisms.
Functional analysis and gene set enrichment analysis (GSEA)
Biological functions of SDEGs were analyzed using the clusterProfiler package, incorporating GO (MF, CC, BP) and KEGG pathways. Additionally, GSEA identified enriched gene sets from hallmark gene sets (h.all.v2022.1.Hs.symbols.gmt) from MSigDB, with FDR < 0.05 indicating statistical significance.
Weighted gene co-expression network analysis (WGCNA) analysis
To identify co-expression modules associated with ischemic stroke, we employed Weighted Gene Co-expression Network Analysis (WGCNA), a systems biology method that clusters genes with similar expression patterns into biologically meaningful modules12. The median absolute deviation (MAD) was calculated for individual genes using their expression profiles, with exclusion criteria applied to the lower 50% of genes demonstrating minimal MAD values. The goodSamplesGenes algorithm from the WGCNA R package was employed to filter out anomalous genes and samples, followed by systematic implementation of scale-free co-expression network construction. Our computational framework initiated with generation of a Pearson correlation matrix through pairwise gene comparisons, subsequently applying average linkage clustering. A weighted adjacency matrix was then formulated through power transformation (a_mn=|C_mn|^β, where C_mn denotes Pearson correlation coefficients), with β operating as a soft-thresholding parameter to amplify significant correlations while attenuating spurious associations.
Upon empirical determination of optimal power (β = 8), topological overlap matrices (TOM) were derived to quantify network connectivity through cumulative adjacency relationships, enabling calculation of gene co-expression ratios and corresponding dissimilarity metrics (1-TOM). Module identification was performed via average linkage hierarchical clustering with TOM-based distance measures, establishing a minimum cluster size threshold of 30 genes. Subsequent refinement with a sensitivity parameter of 3 facilitated dynamic module tree segmentation and eigengene heterogeneity analysis. Final consolidation through pairwise module comparison yielded 23 consensus co-expression modules, with merged modules exhibiting a dissimilarity metric below 0.25.
Immune microenvironment profiling
The CIBERSORT algorithm was applied to determine the relative abundances of 22 distinct immune cell populations within individual specimens. Subsequently, inter-subgroup variations in immune cell infiltration patterns were conducted comparative analyses and graphically represented through bar graph representations. Furthermore, statistically significant associations between central hub genes and specific immune cell subtypes were computed using Pearson correlation coefficients.
Transcriptional regulatory network construction
Potential miRNA and long non-coding RNA (lncRNA) interactions with hub genes were identified through integrated analysis of miRanda, miRDB, and TargetScan databases. Transcriptional regulators associated with the hub genes were predicted using the NetworkAnalyst platform (https://www.networkanalyst.ca), followed by establishment of competing endogenous RNA (ceRNA) networks. Subsequently, the integrated datasets were graphically represented through Cytoscape software to illustrate the regulatory relationships.
Drug prediction and molecular docking
Furthermore, actual hub gene-related medications were identified from the DGIdb (https://dgidb.org/) database, and then molecular docking was performed using AutoDock Vina 1.1.2 software. The docking outcomes were visualized using PyMol 2.5.
Clinical blood specimen collection
To further identify the expression situation of hub genes in the brain, we are prepared to extract the patients’ blood for PCR experiments. We have collected the blood samples of 20 patients who were treated in the First Hospital of Hebei Medical University and were diagnosed with IS based on imaging examinations from October 1, 2024 to December 31, 2024. We also collected the blood samples of 20 patients who were involved in trauma or underwent health check-ups during the same period as controls, detailed patient characteristics are in the Table 1 below.
Ethics declarations
All blood samples were collected with consent from the patients and/or legal guardians. The approval for this research has been obtained from the Human Research Ethics Committee of the First Hospital of Hebei Medical University (No. 20220541). The consent processes and all associated procedures adhered to Chinese national legislation, institutional guidelines, and the Declaration of Helsinki.
Real-time fluorescence quantitative PCR (RT-qPCR)
RT-qPCR was employed to quantify mRNA expression levels of hub genes. Total RNA isolation was performed using the RNAkey Blood Total RNA Extraction Kit (SM141; SEVEN). Complementary DNA synthesis was conducted with the UnionScript First-strand cDNA Synthesis Mix for qPCR (with dsDNase) (SR511; GENESAND). Each RT-qPCR reaction was prepared with a total reaction volume of 20 µL, comprising 10 µL of 2×GS AntiQ qPCR SYBR Fast Mix (Universal) (SQ410, GENESAND), 0.4 µL each of forward and reverse primers, 4 µL cDNA template, supplemented with nuclease-free water to achieve the final volume. All samples underwent triplicate technical replicates. The thermal cycling protocol comprised three primary phases: Initial denaturation at 95 °C for 10 s was conducted, followed by 40 cycles of denaturation at 95 °C for 10 s, primer annealing at 60 °C for 10 s, and elongation at 72 °C for 30 s. Amplification efficiency verification was performed through melt curve analysis during the final dissociation stage. Relative gene expression quantification was determined using the 2^-ΔΔCt computational method, with GAPDH designated as the endogenous reference gene for normalization.In addition, the primers used for this experiment were purchased from Suzhou GENEWIZ Company, all primers were designed using the hg38 genome as a reference. NFE2L2(NM_006164)(Forward tccaagtccagaagccaaactgac, Reverse ggagaggatgctgctgaaggaatc), PTGS2(NM_000963)(Forward aatctggctgcgggaacacaac, Reverse tgtctggaacaactgctcatcacc), GAPDH(NM_002046)(Forward GATTCCACCCATGGCAAATTC, Reverse CTGGAAGATGGTGATGGGATT).
Statistics
All statistical analyses were performed with R software (version 3.5.2) and its integrated packages. The normality of continuous variables was examined through Shapiro-Wilk testing. Inter-group comparisons utilized independent t-tests (for normally distributed data) or Mann-Whitney U tests (for non-parametric distributions) for independent sample analyses, while multi-group comparisons were implemented using one-way ANOVA with Tukey’s post hoc adjustment. Variable associations were quantified through Pearson’s correlation coefficients (linear relationships) or Spearman’s correlation coefficients (non-linear/non-normal distributions). Statistical significance was established as two-tailed p < 0.05, with false discovery rates controlled via Benjamini-Hochberg adjustment. All effect estimates were presented with 95% confidence intervals (95% CI). Data visualization was performed using ggplot2 (v3.1.0) to generate standardized charts.
Results
Senescence related gene screening for IS
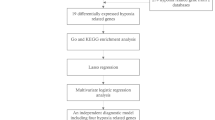
Our technical roadmap is shown in Fig. 1.In this study, we integrated microarray datasets from two Gene Expression Omnibus (GEO) repositories, specifically GSE22255 and GSE458294, comprising a total of 43 control samples and 89 ischemic samples. Prior to conducting data analysis, we addressed and eliminated batch effects present between the different dataset batches. Post batch effect correction, the data distributions across the datasets exhibited increased consistency (Fig. 2A).The UMAP plot shows that samples from various datasets are clustered and interwoven together, indicating a good removal of batch effects (Fig. 2B). The heatmap shows that the two groups of samples exhibit different gene expression profiles (Fig. 2C). Subsequently, we conducted differential expression analysis on the dataset. When the threshold was set to | logFC |>0.5 and FDR < 0.05, 908 differentially expressed genes were identified, as shown in the volcano plot (Fig. 2D). These genes were intersected with the senescence related gene set to obtain 132 senescence related gene sets (Fig. 2E).

Technology flowchart.

Data processing and analysis of GSE22255 and GSE458294 datasets. (A and B) Batch effects were eliminated for the two datasets. (C) Heatmap of gene expression profiles of the two groups of samples. (D) Volcano map of differential expression analysis results. (E) Venn diagram of the intersection of senescence related genes (SGs) and DEGs.
Enrichment analysis and PPI network analysis of the selected SRGs
To further elucidate the potential enrichment functions of the selected SRGs, we conducted a comprehensive Gene Ontology (GO) analysis and a KEGG enrichment study involving 132 genes. As illustrated in Fig. 3A, significant signaling pathways were identified across three categories: molecular function (MF), cellular component (CC), and biological process (BP). Additionally, Fig. 3B demonstrates that the KEGG analysis13 indicated a notable enrichment of differentially expressed genes (DEGs) within the NF-κB signaling pathway, HIF-1 signaling pathway, TGF-β signaling pathway, and various metabolic pathways. The results of the Gene Set Enrichment Analysis (GSEA) (Fig. 3C)further suggest significant involvement of heme metabolism, inflammatory response, and TNFα signaling via NF-κB. The DEGs are analyzed using PPI networks containing 684 pairs of interactions and 102 shared genes based on 132 genes associated with senescence and IS. The networks were visualized using Cytoscape. The result is shown in Fig. 3D.

Functional analysis and gene set enrichment analysis (GSEA) (A) Gene Ontology (GO) analysis (B) KEGG enrichment study (C) Gene Set Enrichment Analysis (GSEA) (D) Network analysis of DEGs.
Implementation of WGCNA methodology
Weighted Gene Co-Expression Network Analysis (WGCNA) methodology was implemented to elucidate gene clusters implicated in Chronic Idiopathic Spontaneous (CIS) pathogenesis. A soft-thresholding power (β = 8) was determined to establish scale-free network topology (Fig. 4A and B). To choose the soft-thresholding power (β),we calculated the scale free topology model fit (R2) and mean connectivity for powers ranging from 1 to 30 (Fig. 4B). The optimal β was selected as the lowest power where R2 exceeded 0.85 (indicating scale-free topology attainment). At β = 8, R2 reached 0.86 while maintaining ideal mean connectivity(30.32) (Fig. 4A). This balanced network robustness against spurious edge inflation. Subsequent hierarchical clustering analysis revealed 23 distinct co-expression modules (Fig. 4C). Through comprehensive module-trait relationship assessment (Fig. 4D), the darkolivegreen module demonstrated the strongest association with CIS phenotype (Pearson’s r = 0.42, P = 6.3 × 10−3; Fig. 4E). Applying rigorous selection criteria (|Module Membership (MM)| > 0.8), this module was classified as the pivotal network hub, encompassing 57 candidate genes with statistically significant expression patterns.

Weighted Gene Co-Expression Network Analysis (WGCNA) (A and B) Scale-free topology (C)WGCNA identification results (D) Correlation analysis (E) Pearson correlation coefficient.
Hub gene selected
We integrated the hub genes identified through Weighted Gene Co-expression Network Analysis (WGCNA) with differentially expressed genes associated with aging, resulting in the identification of seven key senescence related differentially expressed genes(Fig. 5A). Subsequently, we visually delineated the genomic coordinates of the seven differentially expressed genes on their respective chromosomal positions (Fig. 5B). The intergenic associations among these candidate genes were graphically represented in the corresponding subsequent panel (Fig. 5C).

Hub gene selected (A) Venn diagram of the senescence related differentially expressed genes(sDEGs) and genes screened by WGCNA (B) The locations of the 7 DEGs on the chromosomes (C) The relationship among these seven genes.
Machine learning
Subsequently, three machine learning techniques were employed to identify key genes: the LASSO regression algorithm, which revealed four hub genes in the optimization model (Fig. 6A and B); the SVM-RFE algorithm, which selected four hub genes (Fig. 6C); and the RF algorithm, which ranked the top six genes (Fig. 6D and E). Finally, these candidate hub genes were analyzed through a Venn diagram (Fig. 6F), resulting in the identification of three hub genes: GOS2, PTGS2, and ZFP36.

Three machine-learning algorithms are employed for the analysis of hub sDEGs. (A) The coefficient profiles of the least absolute shrinkage and selection operator (LASSO) for the sDEGs. (B) LASSO is utilized for 10 -fold cross-validation to select the optimum tuning parameter. (C) The support vector machine recursive feature elimination (SVM-RFE) estimates the 10 -fold cross-validation error. (D) The Random Forest (RF) algorithm reveals the relationship between the number of decision trees and the model error. (E) The RF algorithm presents the rank of importance of the sDEGs. (F) The Venn diagram indicates that hub sDEGs are screened out through the above three algorithms.
Validated in the GSE16511 gene set
We assessed the expression levels of three genes within the integrated dataset. Relative to the normal group, the ischemic group exhibited a significant upregulation in gene expression (Fig. 7A). To quantitatively evaluate the diagnostic and predictive potential of the optimal gene, we conducted Receiver Operating Characteristic (ROC) curve analysis (Fig. 7B). The Area Under the Curve (AUC) values for the ROC curves were 0.760 for GOS2, 0.758 for ZFP36, and 0.708 for PTGS2, suggesting that these genes possess substantial diagnostic value for ischemic events. Subsequently, we constructed a clinical prognostic model implemented through the ‘lrm’ function within the ‘rms’ package, aimed at assessing the association between PTGS2 expression levels and ischemic stroke risk, with the dose-response relationship visually represented via a dose-response curve (Fig. 7C).External validation of candidate genes was performed using the dataset GSE16511. Relative to the normal control group, PTGS2 was the only gene significantly upregulated (Fig. 7D) and demonstrated diagnostic efficacy for ischemic events. The area under the receiver operating characteristic (ROC) curve (AUC) was 0.788 (Fig. 7E).We also illustrated a model through a line graph to evaluate the association between PTGS2 and the risk of IS (Fig. 7F).

(A) Expression levels of the 3 genes in GSE22255 (B) Receiver Operating Characteristic (ROC) curve analysis of the 3 genes in GSE22255(C) Nomogram of the 3 genes in GSE22255 for predicting IS.(D) Expression levels of the 3 genes in GSE458294 (E) Receiver Operating Characteristic (ROC) curve analysis of the 3 genes in GSE458294 (F) Nomogram of the 3 genes in GSE458294 for predicting IS.
Single gene analysis of PTGS2
PTGS2 may represent a new candidate gene in IS.To investigate the impact of PTGS2 on the ischemic process, we stratified the transcriptomic data from ischemic patients into high and low expression groups based on PTGS2 expression levels and performed a differential analysis. A heatmap (Fig. 8A) was generated to illustrate the 20 genes exhibiting the highest levels of upregulation and the 20 genes with the most significant downregulation in the comparison between these two expression groups. Employing threshold criteria of |log fold change (logFC)| > 0.5 and P < 0.05, the volcano plot (Fig. 8B) identified 418 upregulated and 73 downregulated genes. Functional enrichment analysis of differentially expressed genes demonstrated that PTGS2 was most significantly enriched in the IL-17 signaling pathway-related biological processes (Fig. 8C). Furthermore, Gene Set Enrichment Analysis (GSEA) revealed PTGS2 plays a key role in interferon-gamma response, inflammatory regulation, and TNFα signaling mediated through the NF-κB pathway (Fig. 8D).

Single gene analysis of PTGS2 (A) Heatmap of the top 20 DEGs between PTGS2 high and low expression group (B) volcano plot of differential expression analysis results (C) Enrichment analysis of DEGs (D) GSEA.
Immune cell infiltration results
Based on the CIBERSORT immune profiling results, the proportions of activated CD4 memory T cells, follicular helper T cells, and monocytes were observed to be higher in comparison to the control samples. In contrast, memory B cells, naive CD4 T cells, resting CD4 memory T cells, and resting mast cells showed reduced proportions compared to control samples (Fig. 9A). Correlation analysis of immune cell subpopulations in ischemic stroke revealed that memory B cells exhibited significant inverse associations with both activated and resting memory CD4 + T cells. Resting mast cells demonstrated marked positive correlations with naive CD4 + T cells and monocytes, while monocytes displayed negative correlations with activated memory CD4 + T cells (Fig. 9B). The expansion of activated CD4 + memory T cells and monocytes signifies enhanced neuroinflammation – a hallmark of both stroke and cellular senescence14. These cells amplify oxidative stress through NADPH oxidase activation, accelerating neuronal senescence via p16INK4a/p21 pathways15. The reduction in naive CD4 + T cells and memory B cells parallels immunosenescence phenotypes in aged hosts, impairing adaptive immunity and increasing infection risk post-stroke16. Importantly, PTGS2 expression exhibited a positive correlation with activated mast cells and a negative association with their resting counterparts (Fig. 9C). Studies17 have shown that upregulation of PTGS2 directly activates mast cells to release matrix metalloproteinases (MMPs) by promoting the synthesis of prostaglandin E2 (PGE2), thereby destroying the blood-brain barrier and ultimately leading to secondary brain damage.

(A) The analysis of the immune microenvironment (B) Correlation analysis of immune cells (C) Relationship between immune cells and expression of PTGS2.
TFmRNA, mirna‒mrna, and CeRNA network construction
We conducted a comprehensive investigation into the regulatory relationship of PTGS2, identifying a total of 13 microRNAs (miRNAs) associated with PTGS2 through searches of the miRanda, miRDB, and TargetScan databases. Additionally, we performed further analysis on 31 long non-coding RNAs (lncRNAs) related to PTGS2. The resulting data were visualized using Cytoscape software to construct a diagram illustrating the regulatory relationships, as depicted in Fig. 10A.Subsequently, we accessed the Network Analyst website (https://www.networkanalyst.ca/). The analysis identified a total of 43 transcription factors involved in the regulation of PTGS2. The corresponding regulatory network diagram is presented in Fig. 10B.

(A) lncRNA-miRNA‒mRNA relationship pair intersection. (B)TF-mRNA relationship pair intersection.
The expression partten of NFE2L2 in IS
We conducted an investigation into the correlation between PTGS2 expression and the levels of transcription factors in ischemic datasets. As illustrated in Fig. 11A, the correlation between PTGS2 and transcription factors is presented for datasets GSE22255 and GSE58294, while Fig. 11B depicts the correlation for dataset GSE16561. Our analysis revealed a significant positive correlation between the expression levels of the transcription factor NFE2L2 and PTGS2 in both datasets.Subsequently, we conducted an in-depth investigation into the diagnostic utility of NFE2L2 in patients with ischemic conditions. The area under the receiver operating characteristic (ROC) curve (AUC) was 0.684 in the GSE22255 and GSE58294 datasets(Fig. 11C), and 0.799 in the GSE16561 dataset(Fig. 11D). Additionally, we observed a positive correlation of NFE2L2 expression in both datasets. Of particular note, transcript abundance of NFE2L2 demonstrated a significant elevation in the ischemic cohort when contrasted with controls, a pattern consistently observed across all analytical datasets (Fig. 11E).

(A) Association of PTGS2 expression with transcriptional regulators’ abundance in ischemic cohorts GSE22255 and GSE58294. (B) Association of PTGS2 expression with transcriptional regulators’ abundance in ischemic cohort GSE16561. (C) Diagnostic efficacy evaluation of NFE2L2 using Receiver Operating Characteristic (ROC) curves in GSE22255 and GSE58294. (D) Diagnostic efficacy evaluation of NFE2L2 using ROC curves in GSE16561. (E) Comparative analysis of NFE2L2 expression profiles across multiple datasets (GSE22255/GSE58294 and GSE16561) within the ischemic transcriptional landscape.
Drug prediction and molecular docking of NFE2L2-PTGS2 drug gene interaction analysis
Based on the aforementioned analysis, our findings indicate that NFE2L2-PTGS2 plays a significant role in the pathogenesis and progression of ischemia, suggesting its potential as a therapeutic target. Utilizing the Drug-Gene Interaction Database (DGIdb), we identified several pharmacological agents capable of concurrently targeting NFE2L2(Fig. 12A) and PTGS2(Fig. 12B). We analyzed these agents through a Venn diagram (Fig. 12C), resulting in nine pharmacological agents capable of concurrently targeting NFE2L2.Among these, etodolac exhibited the highest interaction score. Etodolac, a nonsteroidal anti-inflammatory drug (NSAID) and a selective COX-2 inhibitor, is known for its established anti-inflammatory and neuroprotective properties. It is currently utilized in the treatment of inflammation and pain associated with osteoarthritis and rheumatoid arthritis18. Molecular docking performed with AutoDock Vina 1.1.2 revealed the following results: a binding energy of -7.8 kcal/mol. Key interactions included hydrogen bonds formed with the PTGS2 active site residues Arg120 and Tyr355 (bond lengths of 1.9 Å and 2.2 Å, respectively), as well as hydrophobic interactions with Val349 and Leu352. These metrics suggest high-affinity binding, consistent with etodolac’s known mechanism of suppressing prostaglandin synthesis in inflammatory pathways19. Fig. 12D illustrates the molecular docking configuration of PTGS2 with etodolac.

(A)Pharmacological agents capable of targeting PTGS2 (B) Pharmacological agents capable of targeting NFE2L2. (C) Venn diagram of pharmacological agents capable of targeting PTGS2 and NFE2L2 (D) Molecular docking of PTGS2 with etodolac.
Verification of hub senescence-related genes expression by RT-qPCR
Through previous analyses, we have identified that PTGS2 and NFE2L2 are strongly associated with IS. We further verified the expression of the 2 hub genes in the IS patients by RT-qPCR. Clinical validation was performed in a cohort of 20 acute ischemic stroke patients and 20 matched controls. As summarized in Table 1, stroke patients exhibited significantly higher rates of vascular risk factors (hypertension: 80% vs. 25%, p < 0.001; diabetes: 45% vs. 15%, p = 0.03), confirming typical stroke population characteristics. No significant age or sex differences existed between groups (p > 0.05). Consistent with the analytic predictions, our results showed that the expression levels of these genes in the blood of IS patients are significantly higher than normal levels(Fig. 13).

RT-qPCR results (A) The expression levels of two hub genes. (B) The correlation between NFE2L2 expression and PTGS2 expression. ***=p < 0.001.
Discussion
Ischemic stroke (IS) represents a significant public health concern due to its high incidence and associated morbidity, affecting approximately 11 million individuals globally each year1,2. The condition imposes a substantial healthcare burden, particularly during the recovery phase, which often requires extensive rehabilitation and incurs considerable medical costs1,20. Current therapeutic strategies primarily focus on urgent interventions and rehabilitation, yet they are constrained by limitations such as the critical time window for treatment, variable efficacy, and potential side effects3,21. Hence, there is an urgent need to identify novel biomarkers and therapeutic targets to enhance the management of IS.
This study aims to elucidate the role of age-related gene expression profiles in the context of ischemic stroke, leveraging multi-omics data integration and comprehensive analytical methodologies. Previous investigations have demonstrated a strong correlation between aging and the pathophysiology of IS, indicating that age-related genes are pivotal in the disease’s mechanisms22,23. Our findings reveal 132 differentially expressed genes associated with aging, suggesting their potential as novel biomarkers and therapeutic targets. This research not only enhances our understanding of the molecular underpinnings of IS but also lays the groundwork for future clinical applications and therapeutic interventions.
The analysis of gene expression in our study revealed a significant identification of 132 senescence related differential expression genes among the 908 overall differentially expressed genes. This finding is particularly critical as it underscores the potential of these senescence related genes as biomarkers for ischemic stroke. The distinct expression patterns observed between the ischemic stroke and control groups suggest that these genes may play a pivotal role in the pathophysiology of ischemic stroke. Future directions could explore the translational potential of these genes into clinical settings. The pathway from identifying these genes to their application in clinical diagnostics or therapeutics remains a challenge that necessitates further investigation. Moreover, understanding how these genes interact with known stroke markers could yield valuable insights into synergistic mechanisms involved in stroke pathology, thereby enhancing our capability for early diagnosis and intervention strategies24.
Our pathway analysis demonstrated significant enrichment of the NF-κB, HIF-1, and TGF-β signaling pathways among the identified senescence related genes. The involvement of these pathways in ischemic stroke is well-documented, with NF-κB signaling being crucial for inflammatory responses, which are known to exacerbate ischemic injury25,26,27. The activation of HIF-1 is associated with the cellular adaptation to hypoxia, a common feature of ischemic conditions, and may also influence angiogenesis post-stroke28. Similarly, the TGF-β pathway has been implicated in tissue repair mechanisms and fibrosis post-injury29,30. Emerging evidence suggests that NF-κB and HIF-1α synergistically activate PTGS2 under hypoxic and inflammatory conditions, amplifying COX-2-mediated prostaglandin synthesis and senescence-associated secretory phenotype (SASP)31. Conversely, NFE2L2 (encoding NRF2) antagonizes TGF-β signaling by suppressing ROS accumulation, thereby mitigating TGF-β-driven fibroblast senescence32. Understanding how these pathways intersect with senescence related gene expression could provide new avenues for therapeutic intervention. Future studies should focus on elucidating the specific roles of these pathways in mediating stroke outcomes and how modulation of these pathways could alter disease progression or recovery.
The immune profiling conducted via CIBERSORT revealed significant alterations in immune cell populations in ischemic stroke patients, notably an increase in activated CD4 memory T cells and monocytes, alongside a decrease in memory B cells. This shift in immune cell composition not only highlights the immune system’s response to ischemic events but also suggests potential therapeutic targets for immunomodulation strategies. The role of T cells and monocytes in neuroinflammation following ischemic stroke is particularly noteworthy, as they may contribute to both detrimental inflammatory responses and beneficial reparative processes. Investigating the dynamics of these immune cells and their interactions could yield insights into the immunological aspects of stroke recovery and the development of targeted immunotherapeutic approaches. Furthermore, understanding how these immune changes correlate with aging could enhance patient stratification and treatment personalization in clinical settings.
The identification of nine drugs capable of concurrently targeting NFE2L2 and PTGS2 offers promising avenues for pharmacological intervention in ischemic stroke. Notably, etodolac emerged with the highest interaction score, which warrants further exploration into its therapeutic efficacy in this context. As a COX-2 inhibitor, etodolac reduces neuroinflammation by blocking prostaglandin E2 (PGE2) synthesis, thereby attenuating blood-brain barrier disruption and neutrophil infiltration17. The dual-targeting strategy could provide a more comprehensive approach to mitigating the pathological processes associated with stroke. Given that both NFE2L2 and PTGS2 are involved in inflammatory responses, understanding how these drugs modulate their expression and activity could enhance treatment outcomes. This aligns with recent literature advocating for drug repurposing as a viable strategy in addressing acute stroke therapies33,34. Future research should prioritize clinical trials that assess the safety and efficacy of these drugs in stroke patients, particularly in relation to their impact on the identified senescence related pathways and their associated clinical outcomes.
Our analysis revealed a significant positive correlation between the transcription factor NFE2L2 and PTGS2, suggesting a potential regulatory relationship that may influence the pathophysiology of ischemic stroke. The upregulation of NFE2L2 in stroke patients indicates its potential role as a mediator of oxidative stress responses, which are critical in ischemic injury35. Investigating the precise functional interplay between NFE2L2 and PTGS2 could elucidate how these factors contribute to the inflammatory milieu observed in strokes. The characterization of this relationship could also pave the way for targeted therapies aimed at modulating their activity in ischemic conditions. Future studies should explore the mechanistic pathways linking these two factors and how they affect downstream effects on neuroinflammation and tissue repair post-stroke, ultimately contributing to more effective therapeutic strategies.
This study is not without its limitations, which must be acknowledged to contextualize the findings. Firstly, the lack of validation through wet lab experiments raises concerns about the reproducibility and biological relevance of the identified senescence related genes and their proposed mechanisms. Furthermore, the relatively modest sample size may limit the generalizability of the results, as larger cohorts could reveal additional insights or confirm the observed associations. Additionally, potential batch effects among the datasets utilized may influence the reliability of the differential expression analyses, thus warranting caution when interpreting the results. The absence of clinical validation further compromises the translational potential of the identified biomarkers and therapeutic targets, highlighting the need for future studies to incorporate diverse populations and experimental approaches to substantiate these findings.
In conclusion, this research has successfully identified 132 senescence related genes associated with ischemic stroke, providing valuable insights into their roles within various biological pathways and the immune microenvironment. The implications of these findings extend to the identification of potential therapeutic targets, which may enhance treatment strategies for ischemic stroke. Despite the acknowledged limitations, the results lay a foundation for future investigations aimed at elucidating the complex interplay between aging and ischemic stroke. The exploration of these gene interactions and their clinical applications will be crucial in advancing personalized medicine approaches for stroke management, ultimately improving patient outcomes.
Data availability
In this study, RNA expression profiles and clinical information from the stroke datasets GSE16561, GSE22255, and GSE58294 were retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) by the keywords “ischemic stroke”.
References
Feske, S. K. Ischemic Stroke (American Journal of Medicine, 2021).
Walter, K. What is acute ischemic stroke. JAMA 327(9), 885–885. (2022).
Mosconi, M. G. Treatments in ischemic stroke: current and future. Eur. Neurol. 85 (5), 349–366 (2022).
Pu, L., Zhang, L. W. R. & Jiang, T. Z. Y. Projected global trends in ischemic stroke incidence, deaths and disability-adjusted life years from 2020 to 2030. Stroke 54 (5), 1330–1339 (2023).
Q Ding, S. L., Yao, Y. & Cai, H. L. T. Global, regional, and National burden of ischemic stroke, 1990–2019. Neurology 98 (3), e279–e290 (2022).
Tuohy, M. C. & Marshall, E. H. The age-dependent immune response to ischemic stroke. Curr. Opin. Neurobiol. 78, 102670 (2023).
Bretzner, M. & Schirmer, A. K. B. M. D. Radiomics-derived brain age predicts functional outcome after acute ischemic stroke. Neurology 100 (8), e822–e833 (2023).
Wu, Q., Zhang, Y. N. Z. N. N. & Cai, Q. Y. L. Age affects the association of red blood cell indices with efficacy of remote ischemic conditioning in patients with acute moderate ischemic stroke. Sci. Rep. 14 (1), 22561 (2024).
Candelario-Jalil, E. Impact of aging and comorbidities on ischemic stroke outcomes in preclinical animal models: a translational perspective. Exp. Neurol. 335, 113494 (2021).
Ekkert, A., Grigaitė, A. Š. J. & Utkus, B. B. Ischemic stroke genetics: what is new and how to apply it in clinical practice. Genes 13 (1), 48 (2021).
Gasull, T. Molecular mechanisms and pathophysiology of acute stroke: emphasis on biomarkers in the different stroke subtypes. Int. J. Mol. Sci. 23 (16), 9476 (2022).
Abed, S., Ebrahimi, A., Fattahi, F., Shekari-Khaniani, M. & Mansoori Derakhshan, S. Revolutionizing alzheimer’s detection: Immune-Related gene biomarkers as Non-Invasive predictors. Mol. Neurobiol. : 1–12. (2025).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Lian, Z. et al. CD4 + T cells in ischemic stroke: effects and therapeutic targets. Front. Immunol. 16, 1512634 (2025).
Wagner, K. & Wagner, N. The senescence markers p16INK4A, p14ARF/p19ARF, and p21 in organ development and homeostasis. Cells 11, 1966 (2022).
Pangrazzi, L. & Weinberger, B. T cells, aging and senescence. Exp. Gerontol. 134, 110887 (2020).
Li, R. et al. LPS-induced PTGS2 manipulates the inflammatory response through trophoblast invasion in preeclampsia via NF-κB pathway. Reprod. Biol. 22, 100696 (2022).
Gaddala, P. et al. Etodolac utility in osteoarthritis: drug delivery challenges, topical nanotherapeutic strategies and potential synergies. Therapeutic Delivery. 15, 977–995 (2024).
Pentu, N., Azhakesan, A. & Kumar, P. K. Insilico molecular Docking and ADME/T studies of flavonol compounds against selected proteins involved in inflammation mechanism. J. Appl. Pharm. Res. 13, 95–111 (2025).
Wang, M., HQ Gu, C. J. W. & Jiang, X. M. Y. Sex differences in short-term and long-term outcomes among patients with acute ischemic stroke in China. Stroke 53 (7), 2268–2275 (2022).
Xiong, Y. & Fisher, A. K. W. Advances in acute ischemic stroke therapy. Circ. Res. 130 (8), 1230–1251 (2022).
Finger, C. E. & Gutierrez, I. M. Age-related immune alterations and cerebrovascular inflammation. Mol. Psychiatry. 27 (2), 803–818 (2022).
Mechtouff, L. & Crespy, L. R. A narrative review of the pathophysiology of ischemic stroke in carotid plaques: a distinction versus a compromise between hemodynamic and embolic mechanism. Annals Translational Med. 9 (14), 1208 (2021).
FA Montellano, K. U. & Ramiro, L. Role of blood-based biomarkers in ischemic stroke prognosis: a systematic review. Stroke 52 (2), 543–551 (2021).
Li, R., Zhang, Y. Z. S. & Zheng, J. L. Y. The natural (poly) phenols as modulators of microglia polarization via TLR4/NF-κB pathway exert anti-inflammatory activity in ischemic stroke. Eur. J. Pharmacol. 914, 174660 (2022).
Xian, M., Zheng, J. C. K., Liu, Q. L. Y. & Liang, H. L. Aloe-emodin prevents nerve injury and neuroinflammation caused by ischemic stroke via the PI3K/AKT/mTOR and NF-κB pathway. Food Funct. 12 (17), 8056–8067 (2021).
Ran, Y., Gao, W. S. F. & Yang, Z. D. Curcumin ameliorates white matter injury after ischemic stroke by inhibiting microglia/macrophage pyroptosis through NF-κB suppression and NLRP3 inflammasome Inhibition. Oxid. Med. Cell. Longev. 30:2021, 1552127 (2021).
Vatte, S. HIF-1, an important regulator in potential new therapeutic approaches to ischemic stroke. Neurochem Int. 170, 105605 (2023).
Zhang, L., Ai, W. W. X. & Hermann, E. K. Extracellular vesicles from hypoxia-preconditioned microglia promote angiogenesis and repress apoptosis in stroke mice via the TGF-β/Smad2/3 pathway. Cell Death Dis. 12 (11), 1068 (2021).
Li, J., Nie, N. W. H., Jiang, S. W. T. & Liu, X. M. Long non-coding RNA RMST worsens ischemic stroke via microRNA-221-3p/PIK3R1/TGF-β signaling pathway. Mol. Neruobilogy. 59 (5), 2808–2821 (2022).
Horváth, E., Sólyom, Á., Székely, J., Nagy, E. E. & Popoviciu, H. Inflammatory and metabolic signaling interfaces of the hypertrophic and senescent chondrocyte phenotypes associated with osteoarthritis. Int. J. Mol. Sci. 24 (22), 16468 (2023).
Peng, F. et al. 2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis. Signal. Transduct. Target. Therapy. 9, 133 (2024).
Wu, Q., Liu, C. C. W. & Weng, Y. Z. G. Network-based drug repurposing for potential stroke therapy. Comput. Struct. Biotechnol. J. 21, 2809–2823 (2023).
D Ghosh, K. S. & Sodnar, B. Drug repurposing for stroke intervention. Drug Discovery. 27 (7), 1974–1982 (2022).
Liu, C., Tao, B. W. Y., Lou, X. L. X. & Guo, Z. W. Identification and immunological characterization of cuproptosis-related molecular clusters in ischemic stroke. Neuroreport 35 (1), 17–26 (2024).
Funding
This work is founded by National Health Commission Capacity Building and Continuing Education Center-funded project(GWJJQ2022100102).
Author information
Authors and Affiliations
Contributions
Nanding Li and Shanquan Jing conceived the study design, conducted literature review, and wrote the original manuscript draft. Wei Liu and Conghui Li supervised the research framework, provided critical revisions for intellectual content, and secured funding acquisition. Kang An performed data curation and formal analysis, while Bo Jia designed experimental protocols and validated methodological approaches. Yingjie Han contributed to conceptualization and cross-verified theoretical interpretations. Zan Wang developed computational tools and assisted in software implementation. Chengcai Li conducted result verification and comparative benchmarking. Xiaole Li executed statistical analyses and visualized complex datasets. Jiwei Wang provided technical expertise in domain-specific interpretations. All authors critically reviewed the manuscript, approved the final version for publication, and agreed to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, N., Jing, S., An, K. et al. Analyses of the mechanism and therapeutic targets of senescence related genes in ischemic stroke with multi-omics approach. Sci Rep 15, 26178 (2025). https://doi.org/10.1038/s41598-025-10367-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10367-6
