
Abstract
Non-fullerene acceptors (NFAs) play a crucial role in enhancing the performance of bulk heterojunction organic solar cells (BHJ-OSCs). Therefore, new A–π–D–π–A configured NFAs (BDTD1-BDTD7) were developed from BDTR reference through terminal modification with benzothiophene (BT) based acceptors. Density functional theory (DFT) and time-dependent DFT (TD-DFT) methods were employed at M06/6-311G (d, p) level to explore the geometrical, electronic, optical and photovoltaic properties of the designed derivatives. Lower energy gaps (Egap= 2.130–2.250 eV) and higher absorption wavelengths (λmax = 731.249-775.672 nm in chloroform solvent) were obtained for these chromophores. Least values of binding energy (Eb= 0.528–0.554 eV) showed significantly high rate of exciton dissociation which indicated more charge transfer in BDTR and BDTD1-BDTD7. Further, the transition density matrix (TDM), hole-electron and density of states (DOS) graphs also supported the significant charge transfer within titled chromophores. Among all candidates, BDTD5 revealed the best results i.e., least energy gap (2.130 eV) and the bathochromic absorption spectra (775.672 nm). The open-circuit voltage (Voc) was calculated through Scharber’s equation by utilizing PTB7-Th donor. All derivatives showed comparable values of Voc with the reference chromophore (BDTR). Thus, it can be concluded that structural modification through the incorporation of BT acceptors is an effective approach for enhancing the optoelectronic and photovoltaic characteristics of the organic compounds.
Similar content being viewed by others

Introduction
Solar cells are well recognized for their crucial role in providing a promising solution to meet global energy demands. They offer a clean and sustainable source of renewable energy, with low maintenance costs and significant environmental benefits1. Organic photovoltaic (OPVs) technologies have become popular in recent years owing to their manufacturing methods, structural layouts, and semiconducting materials2. Moreover, OSCs possess unique characteristics, such as low manufacturing cost, mechanical flexibility, ability to process solutions, and tunable energy levels3,4,5. Over twenty years, fullerene-based solar cells have attracted considerable interest in the solar-cell market. However, certain limitations, such as limited energy level tunability, weaker light absorption in the visible region, and lower electron-accepting ability have prevented the use of fullerene-based solar cells.
Non-fullerene acceptors (NFAs) enhance the solar light-emitting performance and energy efficiency6. They are considered to be the most advanced solar cells owing to their unique characteristics, including large surface area, simple manufacturing, tunable energy level, high transparency, and flexibility7. Currently, NFA-based OSCs are reported with the power conversion efficiency (PCE) up to 19%8,9. Among NFAs, non-fused ring acceptors (NFRAs) are recognized as more reliable candidates for solar cell applications in terms of material design, particularly when compared to fused ring electron acceptors (FREAs) because of their ease of synthesis and lower production costs. However, the FREAs showed complex synthetic procedures and low yields along with high preparation costs, which hindered their commercialization as the OSCs10,11,12,13. An outlook on the NFRAs indicates that despite the aforementioned advantages, researchers are actively pursuing ongoing advancements to achieve notable progress in this area. Moreover, it is believed that a strong correlation between the molecular structure and device performance leads to higher efficiency of the NFRAs.
With the emergence of exceptional A − D−A type small molecule non-fullerene acceptors (SM-NFAs), the PCEs of OSCs have increased significantly in recent years14. Electron-rich central cores coupled with acceptor groups in highly delocalized π-systems are intriguing for two-photon absorption15,16. Many researchers focused on improving the photovoltaic performance of SM-NFAs by modifying their highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and their energy gap (Egap = ELUMO-EHOMO) by virtue of the Koopman’s theorem which estimates the ionization potential (IP) and electron affinity (EA)17. Study shows that material with a low energy gap is advantageous for light absorption and for producing a substantial photocurrent (Jsc)18. Recently, a theoretical study was performed using the DFT approach on the newly designed organic chromophores (SM1-SM4) obtained via acceptor modification in synthesized BT(-2T-DCV-Hex)2 donor molecule and good photovoltaic properties were obtained by the Scharber equation19.
Chao Li and his collaborators synthesized a small band gap NFA named as BDTI-4 F based on carbon-oxygen bridged ladder type electron-donating core was reported good optoelectronic and photovoltaic properties such as ultra-small energy gap (1.23 eV) and red shifted absorption spectra20. By inspiring from these characteristics, in the current study, new NFAs (BDTR and BDTD1-BDTD7) were designed by end capped modifications with extended acceptors which is more efficient than modifying the π-spacer or core units to improve the photovoltaic properties of NFAs19. These compounds were characterized theoretically using the DFT and TD-DFT approaches to calculate their geometrical, optical and photovoltaic characteristics. Interestingly, the designed materials show smaller band gaps and improved open-circuit voltage (Voc), which aids in improving the PCE of solar cell devices.
Computational procedure
The present quantum computational study was performed using the Gaussian 09 software21, and the GaussView 5.022 enabled visualization of the acquired outcomes. The ChemDraw23 was used to sketch the BDTR and BDTD1-BDTD7 molecules. At first, geometrical optimization was accomplished at M06/6-311G(d, p) level24,25 to obtain ground state structures. After that the optimized geometries were utilized to investigate the optoelectronic, and photovoltaic characteristics of entitled chromophores at aforesaid functional. The Avogadro software26 was utilized to interpret molecular orbital energies and their difference as well as the electronic density probability on counter surfaces of molecular orbitals from gaussian log files. Further, global reactivity parameters (GRPs) were calculated by using the molecular orbital energies via Equations S1-S8. The UV-Vis absorption data was obtained using the GaussSum Version 2.027 from Gaussian outputs and spectra were developed with the aid of Origin 8.028 software. The TDM analysis was performed using the Multiwfn 3.7 software29. Some other software; PyMOlyze27, GaussSum27 and Chemcraft30 were also used to obtain data from outputs.
Results and discussion
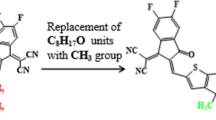
The aim of the current study is to examine the influence of BT acceptors on the optoelectronic properties of the pyran-based NFAs. For this purpose, a synthesized compound (BDTI-4 F) is taken as parent chromophore, consisted of a pyran-based conjugated donor core with thieno[3,4-b]thiophene-2-carboxylate as the π-spacer and (dicyanomethylene)indent-1-one (INCN) as the end groups. It is modified into BDTR, denoted as the reference compound (see Fig. 1), by replacing the 2-ethylhexyl and octyl carboxylate groups with simple methyl groups to reduce the computational cost. Seven derivatives (BDTD1-BDTD7) are designed from BDTR by modifying its terminal acceptor unit, as shown in Scheme 1. Literature have shown that the PCEs of solar cells are remarkably increased by benzothiophene-based acceptors31,32. They can speed up the charge transfer process, while enhancing the open-circuit voltage (Voc) and short-circuit current density (Jsc) values in the OSCs when linked with electron withdrawing units such as –Cl, –F, –NO2, etc33. The IUPAC names of the newly designed chromophores are listed in Table S1 which correspond to their chemical structures as shown in the Fig. S1.

Design of the reference compound (BDTR) from the parent molecule.

Molecular engineering of studied chromophores with extended acceptors.
Geometry optimization
Geometry optimization is a crucial step in the quantum chemical calculations and in the present case it is computed at the M06/6-311G (d, p) level. The Fig. 2 presents the ball and stick model for their true minima fine structures showing all fragments in every derivative. Their side-view geometries are shown in the Fig. 3 which further aid in depicting their planarity via the dihedral angle calculation. Every individual compound shows three significant regions marked as donor (D), π–spacer and acceptor (A). Their point of attachment is considered important in determining the plane of a molecule. There are four dihedral angles (θ1, θ2, θ3, and θ4) in each individual compound. The θ1 and θ4 are formed between the A and π–spacer, while, the θ2 and θ3 show attachment of π–spacer with central D moiety. The values for these mentioned angles are recorded in the Table S11. All the computed values are negative and lie either close to 180° or 0° which confirmed their planar nature. In case of θ1 and θ4 the dihedral angles range from − 161.261° to -165.846°, while in case of θ2 and θ3 they range from − 11.240° to -21.851°. Moreover, negative values of dihedral angles show high electron delocalization and orbital overlap due to planar molecular configurations34. Similarly, their respective bond lengths (Lc−c) are also calculated in the Table S10 which range from 1.398 to 1.431 Å. They fall between the experimental values of C-C bond (1.54 Å) and C = C bond (1.34 Å). Thus, it shows significant π–conjugation among all the molecular units (D, A and π–spacer)35.


Optimized structures of the reference compound BDTR and chromophores (BDTD1–BDTD6) studied.


Side-view of the optimized structures for the titled compounds (BDTR and BDTD1-BDTD6).
Electronic properties
The molecular orbital energies at ground state optimization geometry (GSOP) and excited state optimization geometry (ESOP) were used to investigate the optical and electrical characteristics of the compounds36. The LUMO is frequently thought of as an electron acceptor, whereas the HOMO is typically thought of as an electron donor. The energy gap (Egap) was defined as ELUMO−EHOMO37. In current study, the M06/6-311G(d, p) level was used to carry out the GSOP and ESOP investigations of the reference (BDTR) and designed chromophores (BDTD1-BDTD7). Table 1 displays the EHOMO calculated at GSOP and ELUMO calculated through ESOP and their energy gap. While, results for other orbitals (HOMO-1/LUMO + 1 and HOMO-2/LUMO + 2) are mentioned in Table S12. Compounds with low Egap exhibit high reactivity, low stability, and high polarizabilities6,38.
Table 1 Shows that the BDTR exhibits estimated values of EHOMO/ELUMO as -5.681 and − 3.526 eV, respectively, along with a corresponding Egap of 2.155 eV. For derivatives (BDTD1-BDTD7), the ELUMO values are obtained as -3.311, -3.340, -3.426, -3.530, -3.602, -3.486 and − 3.560 eV, respectively. While, the EHOMO are found as -5.561, -5.583, -5.614, -5.719, -5.732, -5.663 and − 5.731 eV, respectively. The Egap is considered the most crucial parameter in explaining the optoelectronic and charge transport characteristics of the studied compounds. For BDTD1-BDTD7, it is observed to be 2.250, 2.243, 2.188, 2.189, 2.130, 2.177, and 2.171 eV, respectively, as shown in Table 1. The smallest Egap value is observed for BDTD5 (2.130 eV), owing to the presence of two nitro (− NO2) groups on the aromatic benzene ring of its peripheral acceptor unit. It is observed that the − NO2 group has a negative inductive effect (-I) and is significantly electron-withdrawing. Hence, it allows for easy transfer of the electronic charge from the HOMO towards the LUMO via the π-bridge39. Owing to the extended benzothiophene acceptor units at both ends of the compound (BDTD1), it exhibited the highest energy gap (2.250 eV) among the developed series of NFAs. The incorporation of electron-withdrawing floro (–F) groups at the terminal acceptor moiety in BDTD2 caused a decrease in Egap (2.243 eV) compared to the previous compound (BDTD1)40. Another factor which contributes to this energy gap is the thiophene ring, which is known to improve conjugation and decrease Egap in BDTD240. It is observed that Egap in BDTD3 (2.188 eV) decreases further as compared to BDTD2 due to the replacement of fluoro (–F) with chloro (–Cl) groups in the acceptor unit. The Egap (2.177 eV) of compound (BDTD5) is higher than that of BDTD4, likely due to trifluoromethyl (–CF3) groups at the acceptor. This can be ascribed to the fact that the –CF3 group is less electron-withdrawing than the –NO2 group41. The band gap of BDTD6 (2.171 eV) was further reduced owing to the accommodation of sulfonic acid (–SO3H) groups6. A declining order of ΔE is given in eV as follows: BDTD1 (2.250) > BDTD2 (2.243) > BDTD4 (2.189) > BDTD3 (2.188) > BDTD5 (2.177) > BDTD6 (2.171) > BDTR (2.155) > BDTD5 (2.130).
Figure 4 indicates the electron density over the HOMO and LUMO in all the studied compounds calculated through FMOs analysis. It is noted that for the HOMO, the major electron density is located on the donor region and π-spacers. In the LUMO, the terminal acceptors and π-spacers are rich in electron density. Similar results are seen in contour surfaces of other orbitals mentioned in the Fig. S2. In short, the BDTD5 chromophore shows the least energy gap between orbitals and effective intramolecular charge transfer (ICT) as compared to the other chromophores under study, indicating that it might be a valuable material for the photovoltaic applications.


Contour surface diagrams of BDTR and designed molecules (BDTD1-BDTD7).
Reactivity parameters
The reactivity parameters of the investigated molecules are determined using their FMOs energies. These parameters are computed using the Equations S1-S8, as mentioned in the supplementary information. The calculation of the maximal electron transfer from the nucleophile to the electrophile for any molecule under investigation is made easier using the Equation S842. The chemical and kinetic stabilities of designed compounds were closely correlated with the HOMO/LUMO energy difference (Egap = ELUMO−EHOMO)43,44. A compound with smaller ELUMO and EHOMO energy gap is considered as soft molecule with greater polarizability and hence show higher reactivity and vice vera45.
Table 2 shows the comparable values of IP = 5.561–5.732 eV, EA = 3.311–3.602 eV, and X = 4.436–4.667 eV for all derivatives with reference chromophores. Because of strong end-capped acceptors, these results explain why they have a stronger inclination to take electrons46. Similarly, the chemical hardness (η) and softness (σ) are important for estimating the stability and reactivity of compounds43,47. Molecules with a higher energy gap are assumed to be chemically harder, more stable, and less reactive, while molecules with smaller band gaps are thought to be soft, unstable, and highly reactive compounds48. Compound (BDTD5) has the lowest global hardness (η) of 1.065 eV and the highest softness (σ) value of 0.469 eV− 1 among the designed compounds. In case of chemical potential and electronegativity, the highest negative value is obtained for BDTD5 (-4.667 eV) and the highest electronegative molecule is BDTD5 (4.667 eV). In summary, the molecule (BDTD5) exhibited high reactivity and improved charge transfer capabilities. The softness values are determined in the following descending order: BDTD5 > BDTR > BDTD7 > BDTD6 > BDTD3 > BDTD4 > BDTD2 > BDTD1. The distinctive characteristics of BDTD5 might be due to the existence of strongly electronegative –NO2 units on the end-capped benzothiophene-based acceptor.
Density of States (DOS)
To validate the FMOs analysis, the density of states (DOS) is estimated to analyze the contribution of each fragment to the electronic distribution and total absorption band49. The probability of electronic cloud in studied compounds is investigated using DOS analysis, and the graphs are shown in Fig. 5. For this purpose, BDTR and BDTD1-BDTD7 are fragmented into the acceptor (end-group), donor (core unit), and π-bridge (spacer), as shown by the green, red, and blue lines, respectively. The valence band (HOMO) is shown on the left side of the DOS graphs whereas, the conduction band (LUMO) is depicted on the right side. The distance within the HOMO and LUMO indicates the energy gap50.
Table S13 shows that for BDTR the acceptor contributes 50.6% to LUMO and 17.8% to HOMO, donor shows 20.3% to LUMO and 59.6% to HOMO, while π-spacer contributes 29.0% to LUMO and 22.7% to HOMO. The acceptor contributes 54.7, 54.0, 51.0, 50.2, 49.9, 48.8, and 49.8% to LUMO and 18.1, 15.7, 17.2, 16.7, 17.8, 17.2 and 17.1% to HOMO for BDTD1-BDTD7, respectively. While, donor contributes 19.1, 19.5, 21.1, 20.6, 21.2, 22.1and 20.9% to LUMO and 57.5, 59.9, 58.7, 60.8, 59.7, 59.3, and 60.6% to HOMO for BDTD1-BDTD7, respectively Similarly, the π-spacer contributes 26.2, 26.5, 27.8, 29.2, 28.9, 29.2 and 29.3% to LUMO, while 24.4, 24.4, 24.1, 22.6, 22.5, 23.4 and 22.3% to HOMO for BDTD1-BDTD7, respectively. The probability of charge density for the HOMO is majorly present on the donor and slightly on the π-spacer. However, the green peaks in the DOS maps indicate that for LUMO the electronic cloud is mostly located on the acceptor components. As described by the FMOs analysis, DOS analysis shows strong delocalization of the electronic structure and possibility of high level of charge coherence, particularly close to the end-capped groups, from the center of the core to the end-group acceptor region which illustrated them as good candidate for photovoltaic applications.


Graphical representation of DOS for the investigated chromophores.
Optical properties
The optoelectronic characteristics of the titled compounds were determined using UV-Visible spectral analysis51,52. It can also detect the types of transitions and charge-transfer properties of the studied molecules. The absorption spectra of BDTR and BDTD1–BDTD6 are measured using the M06/6-311G(d, p) functional53. Moreover, this analysis was performed to evaluate the photophysical characteristics and estimated several factors, including maximum absorption (λmax), excitation energy (E), oscillator strength (fos), and molecular orbital contribution. The intermolecular charge transfer rate, contributing configurations, and type of electronic excitations in the systems under investigation are clarified by the useful information provided by UV-visible analysis54,55. Representative values are displayed in Table 3, while Tables S14-S29 show the other results for the studied compounds. Figure 6 shows the absorption spectra of designed compounds in gas and solvent.
Due to strong electron-withdrawing acceptor groups with prolonged conjugation, the designed molecules have less excitation energies than the reference (BDTR)56,57. Extended conjugation and strong electron-withdrawing terminal units cause a greater bathochromic shift in the UV-Visible absorption spectrum58,59. The molecules under investigation with A–π–D–π–A configuration demonstrate distinct optical responses in both the gas and chloroform phases, characterized by high λmax and low E values (see Table 3)58. The reference compound (BDTR), which shows the λmax values of 701.462 and 761.849 nm in gas and chloroform, respectively, lesser than the λmax values of the designed compounds (BDTD1-BDTD7). However, in the gaseous phase, the λmax values for the derivatives are obtained in a range of 679.324-715.756 nm, whereas in the chloroform solvent, they show 731.249-775.672 nm range of values. This rise is due to polarity of chloroform solvent which results in higher λmax values. Polar solvents such as chloroform greatly promote the rate of charge transfer process. Furthermore, owing to the presence of heteroatoms, like oxygen, sulfur and nitrogen in the studied molecular frameworks, their compatibility with polar solvent is enhanced as compared to gas phase60. The BDTD5 molecule shows the highest absorption wavelength in both the gaseous (715.756 nm) and chloroform (775.672 nm) phases among all the derivatives. The strong electron-withdrawing nitro group (–NO2) may be the cause of the high absorption value, since it successfully draws electrons away from the π-spacer and towards the acceptor moiety. The BDTD1 compound shows the smallest λmax value in both the solvent (731.249 nm) and gas phases (679.324 nm). Other chromophores, such as BDTD3, BDTD5, and BDTD6, show values of 754.916, 761.849, and 755.996 nm in the chloroform phase and 701.343, 704.931, and 705.373 nm in the gaseous phase, respectively. Overall, the derivatives in the solvent phase exhibit an increasing order of λmax, as follows: BDTD1 < BDTD2 < BDTD4 < BDTD3 < BDTD6 < BDTD7 = BDTR < BDTD5 and BDTD1 < BDTD2 < BDTD4 < BDTD3 < BDTR < BDTD6 < BDTD7 < BDTD5 in the gaseous phase. Concluding the discussion, it is inferred that structural modification using acceptor species is beneficial in enhancing the UV-Visible absorption capabilities of the proposed organic compounds. Finally, BDTD5 is the most appropriate compound to be obtained for this purpose because it has the qualities needed for effective charge transfer analysis in photovoltaics.

UV-visible absorption spectra of the titled compounds in gas and solvent phases.
Transition density matrix (TDM) analysis
TDM is a precise method used in the OSCs to examine and identify electronic transition routes61. The features and consistency intervals are provided by the TDM for each electronic excitation between the two eigenstates of the compounds. The main function of TDM is to show the intermolecular charge-transfer excitations in organic molecules62. This research clarifies the processes involved in ICT from S0→S1 and the interactions that occur between electron-donating and electron-accepting entities63,64,65. The calculations are performed at the M06 level using Multiwfn 3.7 software29, and the developed heat maps are shown in Fig. 7. Because the contribution of hydrogen atoms to the transitions is small, they are neglected in this study. The multi-colored lines across the TDM graphs represent the division of the charge density areas of different components of a molecule.
For the TDM analysis, the investigated compounds were categorized as acceptor (A), donor (D), and π-spacer units. It is observed that charge density is prominently located over the terminal acceptor units in all the proposed molecules, which demonstrates the efficient charge shift from the central donor towards end-capped acceptors via the π-spacers as illustrated by red, green and yellow spots over a dark back ground. Thus, it can be concluded that in the proposed organic chromophores good ICT occurred which are demonstrated them as an efficient candidate for OSCs.


Graphical representation of TDM plots for the investigated compounds.
Exciton binding energy (E b) analysis
The exciton binding energy (Eb) is the difference between the electrical and optical energy gap66. In small molecules, the Egap is equal to ELUMO-EHOMO, whereas the optical energy gap is mainly the first exciton energy (Eopt). The binding energy (Eb) is defined as the energy required to dissociate the exciton and investigate the Columbic force in the holes and electrons. Theoretically, the Eb of the compounds was calculated using Eq. 167.
where Eb is the binding energy, EL−H is the energy difference between the LUMO and HOMO, and Eopt is the first excitation energy68. The theoretically calculated binding energies are listed in Table 4.
Among the designed chromophores, the highest observed value of binding energy (Eb) was 0.554 eV, for BDTD1; however, BDTD5 shows the lowest value of 0.532 eV. The decreasing order of binding energy for the derivative compounds was found as BDTD1 > BDTD2 > BDTD3 > BDTD6 > BDTD7 > BDTD4 > BDTD5 > BDTR. Therefore, out of all the derivatives, BDTD5 shows the lowest value of Eb with a greater magnitude of exciton dissociation in the excited state (S1), exhibiting a significant photovoltaic response.
Electron-Hole analysis
Electron-hole analysis is an efficient and comprehensive method to investigate the electron-hole mobility in the studied compounds69. It is performed using the Multiwfn 3.7 software29, and the obtained graphs are displayed in the Fig. 8. In the reference chromophore (BDTR), the electron intensity is concentrated over the carbon atoms (C29 and C30) of the thiophene ring in the π-spacer. Interestingly, in all the designed compounds, the electron intensity is maximum at C33 of the π-spacer (5-methyl-2,3-dihydrothiophene), successively moving towards the acceptor moiety. Conversely, a hole is found at C9 of the donor, indicating significant charge transfer from the π-linker to acceptor moiety. In conclusion, the designed molecules (BDTD1-BDTD7), including the reference, seem to be electron-type materials due to the electronic intensity which is found to be maximum in the electronic band compared to the hole intensity in the hole band (see Fig. 8). Therefore, all studied compounds are found to be efficient materials for organic solar cell applications.

Pictorial representation of electron-hole transport analysis of the studied compounds.
Open circuit voltage (V oc)
Voc is used to estimate the function of the OSCs by calculating the highest voltage obtained at zero current70. This is based on the energy difference between the HOMO and LUMO of the donor and acceptor molecules. The following Scharber’s Equation is used to calculate Voc71:
where Voc is the open-circuit voltage,\(\:\:{\text{E}}_{\text{H}\text{O}\text{M}\text{O}}^{\text{D}}\) is the HOMO energy of the donor,\(\:\:{\text{E}}_{\text{L}\text{U}\text{M}\text{O}}^{\text{A}}\) is the LUMO energy of the acceptor, and 0.3 is known as the voltage drop factor72. A larger Voc results in a faster electron transfer rate from the donor HOMO to the acceptor LUMO in the organic chromophores, which leads to increase light-harvesting ability. The donor polymer (PTB7-Th) is well known and is used to prepare the donor: acceptor complex with the designed acceptor molecules (BDTR and BDTD1-BDTD7). The obtained results are shown in the Table S30, which demonstrates the energy gap between the HOMO of PTB7-Th (-5.20 eV)73 and the LUMO of the designed NFAs molecules, as well as the Voc results, whose pictorial representation is shown in Fig. 9.
The differences between EALUMO and EDHOMO for BDTR and BDTD1-BDTD7 are found as 1.68, 1.89, 1.86, 1.78, 1.67, 1.60, 1.72 and 1.64 eV, respectively. Moreover, the designed molecules significant Voc values as shown in the Table S30. The following increasing order is obtained from the calculated Voc results: BDTD5 (1.30) < BDTD7 (1.34) < BDTD4 (1.37) < BDTR (1.38) < BDTD6 (1.42) < BDTD3 (1.48) < BDTD2 (1.56) < BDTD1 (1.59) in volts (V). Based on these outcomes, the proposed organic chromophores are considered to be best candidates for achieving maximum output voltage and high PCE as OSCs.

Open-circuit voltage (Voc) of tilted chromophores with respect to PTB7-Th.
Conclusion
The photophysical, photovoltaic, and optoelectronic characteristics of recently developed A–π–D–π–A type pyran based fullerene-free chromophores (BDTR and BDTD1-BDTD7) are investigated using the quantum chemical techniques. The BT based end-capped substitution in the designed compounds (BDTD1-BDTD7) lead to bathochromic shift (λmax = 731.249-775.672 nm in chloroform and 679.324–715.756 nm in the gas phase) compared with BDTR (λmax = 761.849 in chloroform and 701.462 nm in the gas phase). Interestingly, the derivatives exhibited lower energy gaps (2.130–2.250 eV) than BDTR (2.155 eV). A good ICT is investigated through FMOs, TDM and DOS analyses. Moreover, electron-hole analysis showed that the proposed molecules are electron-type materials with rapid charge-transfer properties. In particular, the molecule (BDTD5) is regarded as the most efficient design for solar cells owing to its significant characteristics like low band gap (2.130 eV), bathochromic shift (715.756 nm in gas and 775.672 nm in chloroform solvent), and the lowest binding energy (0.532 eV). Furthermore, significant photovoltaic responses are observed for the investigated compounds with respect to the HOMOPTB7−Th –LUMOacceptor. Overall, this study demonstrates that effective modification through BT based end-capped acceptors can facilitate the development of new materials with enhanced electrical and optical properties. It is expected that synthesis of the proposed molecules will lead to the development of highly efficient OSCs.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Shafiq, I. et al. The influence of Selenophene π-linker on photovoltaic properties of pyrrole-4,6(5-H)-dione-based chromophores: A quantum chemical study. J. Ind. Eng. Chem. 136, 589–602 (2024).
Yuan, J. et al. Single-Junction organic solar cell with over 15% efficiency using Fused-Ring acceptor with Electron-Deficient core. Joule 3 (4), 1140–1151 (2019).
Khalid, M. et al. Role of extended end-capped acceptors in non-fullerene based compounds towards photovoltaic properties. J. Photochem. Photobiol., A. 448, 115292 (2024).
Liu, Z. et al. Non-fullerene polymer acceptors based on perylene diimides in all-polymer solar cells. Sol. Energy Mater. Sol. Cells. 189, 103–117 (2019).
Qi, Q. et al. Side-chain optimization of perylene diimide-thiophene random terpolymer acceptors for enhancing the photovoltaic efficiency of all-polymer solar cells. Org. Electron. 78, 105616 (2020).
Khalid, M. et al. Exploration of the interesting photovoltaic behavior of the fused benzothiophene dioxide moiety as a core donor with modification in acceptors for high-efficacy organic solar cells. RSC Adv. 12 (45), 29010–29021 (2022).
Abdelkhalk, A. et al. A computational study of thiophene containing small-molecule electron acceptors for non-fullerene organic photovoltaic cells. Mater. Sci. Energy Technol. 6, 137–144 (2023).
Janjua, M. & Chemistry, P. A. Impact of symmetry breaking on the performance of non-fullerene acceptors (NFAs) for photo and thermally stable organic solar cells (OSCs): a DFT-based interrogation and investigation. J. Photochem. Photobiol. A: Chem. 444, 115003 (2023).
Zhu, L. et al. Single-junction organic solar cells with over 19% efficiency enabled by a refined double-fibril network morphology. Nat. Mater. 21(6), 656–663 (2022).
Li, X. et al. Simplified synthetic routes for low cost and high photovoltaic performance n-type organic semiconductor acceptors. Nat. Commun. 10(1), 519 (2019).
Yang, W. et al. Balancing the efficiency, stability, and cost potential for organic solar cells via a new figure of merit. Joule 5(5), 1209–1230 (2021).
Li, N. et al. Analyzing the efficiency, stability and cost potential for fullerene-free organic photovoltaics in one figure of merit. Energy Environ. Sci. 11(6), 1355–1361 (2018).
Yang, X. & Ding, L. J. J. S. Org. semiconductors: Commercialization market. J. Semicond. 42(9), 090201 (2021).
Taouali, W. et al. Developed non-fullerene acceptors with modified BTPT-OD donor core: A DFT and TD-DFT methods to boost organic solar cell performances. Org. Electron. 140, 107226 (2025).
Katan, C. et al. Two-photon transitions in quadrupolar and branched chromophores: experiment and theory. J. Phys. Chem. B. 111(32), 9468–9483 (2007).
Delgado, M. C. R. et al. Tuning the charge-transport parameters of perylene diimide single crystals via end and/or core functionalization: a density functional theory investigation. J. Am. Chem. Soc. 132(10), 3375–3387 (2010).
Koopmans, T. Über die zuordnung von wellenfunktionen und eigenwerten zu Den Einzelnen elektronen eines atoms. Physica 1 (1–6), 104–113 (1934).
Zhang, L. et al. Theoretical investigations on donor–acceptor conjugated copolymers based on naphtho [1, 2-c: 5, 6-c] Bis [1, 2, 5] thiadiazole for organic solar cell applications. J. Phys. Chem. C. 116 (50), 26154–26161 (2012).
Taouali, W. et al. Theoretical design of new small molecules with a low band-gap for organic solar cell applications: DFT and TD-DFT study. Comput. Mater. Sci. 150, 54–61 (2018).
Li, C. et al. Small bandgap non-fullerene acceptor enables efficient PTB7-Th solar cell with near 0 ev HOMO offset. J. Energy Chem. 52, 60–66 (2021).
Frisch, M. et al. Gaussian 09 (Revision A02) (Gaussian Inc. Wallingford CT, 2009).
Nielsen, A. & Holder, A. Gauss View 5.0 (User’s Reference, GAUSSIAN Inc., 2008).
Cousins, K. R. Computer Review of ChemDraw Ultra 12.0 (ACS, 2011).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Andersson, M. P. & Uvdal, P. New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-ζ basis set 6-311 + G (d, p). J. Phys. Chem. A. 109 (12), 2937–2941 (2005).
Hanwell, M. D. et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 1–17 (2012).
O’boyle, N. M. & Tenderholt, A. L. M.J.J.o.c.c. Langner. Cclib: Libr. package-independent Comput. Chem. Algorithms. 29 (5), 839–845 (2008).
OriginPro, Version 8.5; OriginLab Corporation: Northampton, MA, 2010.
Lu, T. F.J.J.o.c.c. Chen. Multiwfn: Multifunctional Wavefunction Analyzer. 33 (5), 580–592 (2012).
Zhurko, G. October, Chemcraft: http://www.chemcraftprog.com22, (2014).
Khan, M. et al. Exploring the influence of end-capped moieties on the photovoltaic properties of Thiazolo [5, 4-d] thiazole based compounds: DFT/TD-DFT approaches. Mater. Sci. Semiconduct. Process. 187, 109126 (2025).
Shafiq, I. et al. Structural modeling of fluorinated Quinoxaline core–based chromophores for efficient photovoltaic materials: a DFT study. J. Phys. Org. Chem. 38 (1), e4663 (2025).
He, L. J. et al. Fine-tuning π-spacer for high efficiency performance DSSC: A theoretical exploration with D – π – A based organic dye. Dyes Pigm. 141, 251–261 (2017).
Taouali, W. & Alimi, K. Optimizing non-fullerene acceptor molecules constituting fluorene core for enhanced performance in organic solar cells: a theoretical methodology. J. Mol. Model. 30 (10), 342 (2024).
Taouali, W. et al. Exploring the impact of cyano substitutions in non-fullerene acceptors for enhanced organic solar cell performance: A DFT and TD-DFT investigation. Comput. Theor. Chem. 115102. (2025).
Hussain, R. et al. Density functional theory study of palladium cluster adsorption on a graphene support. RSC Adv. 10 (35), 20595–20607 (2020).
Janjua, M. R. S. A. et al. Effect of π-conjugation spacer (CC) on the first hyperpolarizabilities of polymeric chain containing polyoxometalate cluster as a side-chain pendant: A DFT study. Comput. Theor. Chem. 994, 34–40 (2012).
Janjua, M. R. S. A. et al. A DFT study on the Two-Dimensional Second-Order nonlinear optical (NLO) response of Terpyridine-Substituted hexamolybdates: physical insight on 2D Inorganic–Organic hybrid functional materials. 2012 4, 705–711 (2012).
Khalid, M. et al. First theoretical framework for highly efficient photovoltaic parameters by structural modification with benzothiophene-incorporated acceptors in Dithiophene based chromophores. Sci. Rep. 12 (1), 20148 (2022).
Khalid, M. et al. Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: A DFT approach. RSC Adv. 11(23), 14237–14250 (2021).
Hussain, R. et al. Molecular engineering of A–D–C–D–A configured small molecular acceptors (SMAs) with promising photovoltaic properties for high-efficiency fullerene-free organic solar cells. Opt. Quantum Electron. 52, 1–20 (2020).
Juma, J. M., S.A.J.J.o.C, R. & Vuai. Computational studies of the thermodynamic properties, and global and reactivity descriptors of fluorescein dye derivatives in acetonitrile using density functional theory. J. Chem. Res. 45 (7–8), 800–805 (2021).
Shafiq, I. et al. Use of benzothiophene ring to improve the photovoltaic efficacy of cyanopyridinone-based organic chromophores: a DFT study. RSC Adv. 14 (18), 12841–12852 (2024).
Ferdowsi, P. et al. Molecular design of efficient organic D-A-π -A dye featuring triphenylamine as donor fragment for application in dye-Sensitized solar cells. ChemSusChem 11 (2), 494–502 (2018).
Aboulouard, A. et al. New non-fullerene electron acceptors-based on Quinoxaline derivatives for organic photovoltaic cells: DFT computational study. Synthetic Metals. 279, 116846 (2021).
Khalid, M. et al. Influence of end-capped modifications in the nonlinear optical amplitude of nonfullerene-based chromophores with a D – π–A architecture: a DFT/TDDFT study. ACS omega. 7(27), 23532–23548 (2022).
Parr, R. G. & Pearson, R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105(26), 7512–7516 (1983).
Khalid, M. et al. Theoretical designing of non-fullerene derived organic heterocyclic compounds with enhanced nonlinear optical amplitude: a DFT based prediction. Sci. Rep. 12(1), 20220 (2022).
Shehzad, R. A. et al. Designing of benzothiazole based non-fullerene acceptor (NFA) molecules for highly efficient organic solar cells. Comput. Theor. Chem. 1181, 112833 (2020).
Khan, M. U. et al. Novel W-shaped oxygen heterocycle-fused fluorene-based non-fullerene acceptors: first theoretical framework for designing environment-friendly organic solar cells. Energy Fuels 35(15), 12436–12450 (2021).
Khan, M. U. et al. First theoretical framework of triphenylamine–dicyanovinylene-based nonlinear optical dyes: structural modification of π-linkers. J. Phys. Chem. C 122(7), 4009–4018 (2018).
Mahmood, A. et al. Computational designing of triphenylamine dyes with broad and red-shifted absorption spectra for dye‐sensitized solar cells using multi-thiophene rings in π-spacer. Bull. Korean Chem. Soc. 36(11), 2615–2620 (2015).
Mahmood, A. et al. First-principles theoretical designing of planar non-fullerene small molecular acceptors for organic solar cells: manipulation of noncovalent interactions. Phys. Chem. Chem. Phys. 21(4), 2128–2139 (2019).
Khan, M. U. et al. First theoretical probe for efficient enhancement of nonlinear optical properties of Quinacridone based compounds through various modifications. Chem. Phys. Lett. 715, 222–230 (2019).
Bourass, M. et al. The photophysical properties and electronic structures of bis [1] benzothieno [6, 7-d: 6′, 7′-d′] benzo [1, 2-b: 4, 5-b′] dithiophene (BBTBDT) derivatives as hole-transporting materials for organic light-emitting diodes (OLEDs). New J. Chem. 43(40), 15899–15909 (2019).
Ans, M. et al. Opto-electronic properties of non-fullerene fused-undecacyclic electron acceptors for organic solar cells. J. Saudi Chem. Soc. 159, 150–159 (2019).
Shafiq, I. et al. Exploration of photovoltaic behavior of benzodithiophene based non-fullerene chromophores: first theoretical framework for highly efficient photovoltaic parameters. J. Mater. Res. Technol. 24, 1882–1896 (2023).
Khalid, M. et al. Efficient tuning of small acceptor chromophores with A1-π-A2-π-A1 configuration for high efficacy of organic solar cells via end group manipulation. J. Saudi Chem. Soc. 25 (8), 101305 (2021).
Ans, M. et al. Spirobifluorene based small molecules as an alternative to traditional fullerene acceptors for organic solar cells. Mater. Sci. Semicond. Process. 94, 97–106 (2019).
Taouali, W. et al. Exploring the impact of structural modifications of Phenothiazine-Based novel compounds for organic solar cells: DFT investigations. Polymers 17 (1), 115 (2025).
Bourass, M. et al. Electronic structures and photophysical properties of carbazole-and thiophene‐based organic compounds used as hole‐injecting layer for organic light‐emitting diodes (OLEDs). Can. J. Chem. 101(5), 2646–2659 (2023).
Ans, M. et al. Designing three-dimensional (3D) non-fullerene small molecule acceptors with efficient photovoltaic parameters. Chem. Select 3(45), 12797–12804 (2018).
Arshad, M. N. et al. Exploration of the intriguing photovoltaic behavior for fused indacenodithiophene-based A–D–A conjugated systems: a DFT model study. ACS omega 7(14), 11606–11617 (2022).
Mahmood, A. et al. Quantum chemical analysis and molecular dynamics simulations to study the impact of electron-deficient substituents on electronic behavior of small molecule acceptors. Comput. Theor. Chem.1204, 113387 (2021).
Shafiq, I. et al. Structural tailoring via end-capped acceptors of thiophene-based C-shaped non-fullerene compounds with A-π-A backbone for the exploration of photovoltaic response. J. Saudi Chem. Soc. 28 (1), 101799 (2024).
Khan, M. U. et al. Designing spirobifullerene core based three-dimensional cross shape acceptor materials with promising photovoltaic properties for high‐efficiency organic solar cells. Int. J. Quantum Chem. 120(22), e26377 (2020).
Köse, M. Evaluation of acceptor strength in thiophene coupled donor–acceptor chromophores for optimal design of organic photovoltaic materials. J. Phys. Chem. A 116(51), 12503–12509 (2012).
Lesar, A. & Milošev, I. Density functional study of the corrosion inhibition properties of 1, 2, 4-triazole and its amino derivatives. Chem. Phys. Lett. 483(4–6), 198–203 (2009).
Liu, Z., Lu, T. & Chen, Q. J. C. An sp-hybridized all-carboatomic ring, cyclo [18] carbon: electronic structure, electronic spectrum, and optical nonlinearity. Carbon 165, 461–467 (2020).
Mehboob, M. Y. et al. Designing N-phenylaniline-triazol configured donor materials with promising optoelectronic properties for high-efficiency solar cells. Comput. Theor. Chem. 1186, 112908 (2020).
Wazzan, N. A DFT/TDDFT investigation on the efficiency of novel dyes with ortho-fluorophenyl units (A1) and incorporating benzotriazole/benzothiadiazole/phthalimide units (A2) as organic photosensitizers with D–A2–π–A1 configuration for solar cell applications. J. Comput. Electron. 18, 375–395 (2019).
Mehboob, M. Y. et al. Quantum chemical design of near-infrared sensitive fused ring electron acceptors containing Selenophene as π‐bridge for high‐performance organic solar cells. J. Phys. Org. Chem. 34(8), e4204 (2021).
Khan, M. U. et al. Designing electron-deficient diketone unit based non-fused ring acceptors with amplified optoelectronic features for highly efficient organic solar cells: a DFT study. molecules 28(8), 3625 (2023).
Acknowledgements
The authors thank the Ongoing Research Funding Program (ORG-2025-6), King Saud University, Riyadh, Saudi Arabia. Further, K.C. acknowledges the support from the doctoral research fund of the Affiliated Hospital of Southwest Medical University.
Author information
Authors and Affiliations
Contributions
Mashal Khan: Conceptualization; Formal analysis; Investigation; Review & editing; Visualization; Funding acquisition.Maria Zafar: Data curation; Visualization; Conceptualization; Formal analysis; Investigation; Writing - original draft; VisualizationZafar Ullah: Data curation; Visualization; Conceptualization; Formal analysis; Investigation; Writing - original draft; VisualizationIqra Shafiq: Conceptualization; Formal analysis; Investigation; Review & editing; Visualization; Funding acquisition.Saifullah Bullo: Conceptualization; Supervision; Formal analysis; Investigation; Review & editing; Resources; software; project administration; MethodologyTansir Ahamad: Conceptualization; Supervision; Formal analysis; Investigation; Validation; VisualizationKe Chen: DFT data acquisition, Review & editing, Analysis, Funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khan, M., Zafar, M., Ullah, Z. et al. Tuning peripheral acceptors in pyran core functional materials to boost photovoltaic efficiency. Sci Rep 15, 26368 (2025). https://doi.org/10.1038/s41598-025-11718-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11718-z
