
Abstract
Emerging evidence suggests that dysregulation of lipid metabolism and chronic inflammation in the central nervous system (CNS) contribute to the risk of Parkinson’s disease (PD). However, the causal relationship between plasma lipid metabolism and PD, particularly through immune modulation, remains unclear. In this study, we used Mendelian randomization (MR) to investigate the causal association between plasma lipidome and PD risk, utilizing GWAS summary statistics for PD, circulating immune cells, inflammatory proteins, and plasma lipidome. We found that higher plasma phosphatidylcholine (17:0_18:1) levels were causally associated with an increased risk of PD, with a 1 standard deviation genetically instrumented higher level corresponding to a 30% increased risk (95% CI 1.05–1.61, p = 0.014). Multivariate Mendelian randomization (MVMR) analysis confirmed this association after adjusting for triacylglycerol levels. Mediation analysis revealed that the causal link between plasma phosphatidylcholine and PD was mediated by circulating immune cells (specifically CD45 on CD14+ monocytes and CD45 on HLA-DR+ T cells) rather than by circulating inflammatory proteins. These findings highlight a novel pathway linking lipid metabolism, immune modulation, and PD risk.
Similar content being viewed by others


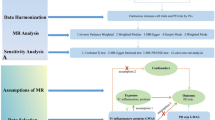
Introduction
Parkinson’s disease (PD) is one of the most prevalent neurodegenerative disorders, primarily characterized by a group of motor symptoms collectively referred to as parkinsonism. These symptoms include tremor, muscle rigidity, bradykinesia (slowness of movement), and disturbances in gait and balance1,2. Epidemiological data indicate that PD affects approximately 1% of individuals over the age of 60, with incidence rates ranging from 1 to 2 cases per 1,000 people in general populations3,4. Although PD is rare before the age of 50, its prevalence rises to 4% in the oldest age groups5. Therefore, PD is also considered the primary cause of disability in middle-aged and older adults globally1,3. Neuropathologically, PD is marked by the presence of α-synuclein (α-Syn) aggregates, known as Lewy bodies, and the degeneration of dopaminergic neurons in the substantia nigra pars compacta6. Currently, several hypotheses, including oxidative stress, mitochondrial dysfunction, lysosomal dysfunction, and inflammation, have been proposed to explain the neurodegeneration observed in PD7,8,9,10. However, the precise causes of PD remain unclear. Recent genetic studies have increasingly supported the hypothesis that PD results from a complex interaction between genetic susceptibility, aging, and environmental factors11,12.
The brain has one of the highest lipid concentrations among body tissues, containing a variety of lipid types, including fatty acids, triacylglycerols, phospholipids, sterols, and glycolipids13. Recent large-scale high-throughput sequencing studies have uncovered several PD risk genes, many of which are linked to lipid-related pathways14,15,16,17,18,19,20,21,22,23,24. The most significant genetic factor associated with PD risk is missense mutations in GBA1, which encodes glucocerebrosidase, a lysosomal enzyme involved in lipid metabolism14,15. Mutations in SMPD1, which encodes acid sphingomyelinase, as well as polymorphisms in GALC and ASAH1, which encode lysosomal enzymes that break down sphingolipids, have also been associated with increased PD risk16,17,18. Other lipid-related genes linked to PD include DGKQ, which encodes diacylglycerol kinase theta19,20, responsible for the regeneration of phosphatidylinositol from diacylglycerol, a key process for synaptic vesicle formation21,22, and SREBF1, which encodes the sterol regulatory element-binding transcription factor 123, a crucial regulator of lipid biosynthesis and membrane integrity24. Advanced high-resolution histochemical techniques have revealed that Lewy bodies contain not only α-synuclein fibrils but also membrane lipids, mitochondria, and lysosomes25, emphasizing the critical role of lipids in the physiological and pathological behavior of α-synuclein13. Additionally, the identification of ASAH1, GBA1, and GALC as genes linked to substantial Lewy body deposition supports the hypothesis that lipids play a key role in the aggregation and propagation of α-synuclein in PD.
Phospholipids are crucial components of cell membranes, vital for maintaining both membrane fluidity and topology26. Due to the dynamic nature of membrane-bound proteins and transporters, phospholipids—and their breakdown products—can influence various cell signaling pathways involved in neuroprotection and neurodegeneration27,28. Phosphatidylcholines (PCs), for instance, are structural lipids that also play key roles in enzyme activation and membrane-mediated cell signaling29. Lysophosphatidylcholine (LPC) is produced under both normal and pathological conditions through the spontaneous hydrolysis or enzymatic breakdown of PCs29,30. Due to their detergent-like properties, LPCs are essential for modulating membrane characteristics31. Additionally, LPCs act as secondary messengers, exerting dual effects by triggering neuroinflammatory signaling and influencing both pro-apoptotic and anti-apoptotic pathways32,33. However, it remains unclear whether these effects are the result of direct action by lipids as second messengers in signal transduction or through the modulation of related enzyme activity.
A growing body of evidence implicates PCs and LPCs in the modulation of diverse disease-related pathways, with reported associations across a spectrum of conditions, including diabetes, obesity, cancer, atherosclerosis, myocardial infarction, and neurological and neurodegenerative disorders34,35. For example, Mulder et al.34 observed no significant differences in total PC levels between patients and controls; however, LPC concentrations were reduced in the cerebrospinal fluid of individuals with Alzheimer’s disease (AD), resulting in a decreased LPC/PC ratio. Similarly, a lower plasma LPC/PC ratio has been reported by Fuchs et al.36 as a potential biomarker for disease severity in rheumatoid arthritis. To date, however, no data are available on the plasma PC/LPC profile in PD, despite the possibility that this ratio could reflect degenerative processes and aid in disease stratification. We therefore sought to investigate a potential causal relationship between plasma PC levels and PD risk using Mendelian randomization analysis.
Method
Traits analyzed and genome-wide association studies
The summary statistics from the genome-wide association study (GWAS) meta-analysis for PD, circulating immune cells and inflammatory proteins, and the plasma lipidome are publicly available. The largest available meta-analysis of individuals of European ancestry provided genetic association data for the plasma lipidome37. In brief, we utilized genotype data from the GeneRISK cohort, which examined genome-wide correlations for 179 lipid species across 7,174 participants. Genetic information on circulating immune cells was sourced from the GWAS summary statistics of Orru et al.38, which included 731 immunophenotypes categorized into four groups: absolute cell counts (n = 118), median fluorescence intensities reflecting surface antigen levels (n = 389), morphological parameters (n = 32), and relative cell counts (n = 192). Data on circulating inflammatory proteins were derived from the GWAS project and assessed across 11 cohorts of 14,824 individuals of European ancestry using the Olink Target Inflammation Panel39. Genetic data on PD were obtained from a cross-population genetic correlation map40. In summary, we performed an inverse-variance meta-analysis on data from individuals of European and East Asian ancestry, adjusting for age, sex, and the main components representing ancestry (2,978 cases; 635,168 controls) (Table 1).
Mendelian randomization analyses
GWAS summary statistics were utilized to conduct bidirectional two-sample Mendelian randomization (MR), with α̂ Lipids → PD representing the causal effect of lipids (exposure) on PD (outcome), and α̂ PD → Lipids indicating the causal effect of PD (exposure) on lipids (outcome). Harmonized SNPs, significantly associated (p < 5 × 10⁻⁸) with the exposure, were clumped (p₁ = 0.0001, p₂ = 0.01, kb = 10,000, and r2 = 0.01) using PLINK v1.941,42,43 and retained as instrumental variables (IVs). SNPs located within the complex long-range linkage disequilibrium (LD) structure of the HLA locus (chr6:25,000,000–37,000,000; GRCh37/hg19) were excluded from the IVs due to known confounding effects44. For each exposure-outcome pair, further IVs were removed based on significant differences in allele frequency (≥ 0.05) and Steiger filtering (Z ≤ − 1.96). Bidirectional MR analyses were conducted using the TwoSampleMR R package (v0.5.7)45, primarily employing the inverse variance-weighted (IVW) method. Complementary MR methods were also applied. Specifically, MR-Egger regression was utilized, as it is considered conservative in the presence of pleiotropic variants and reduces the risk of inflated test statistics that could lead to false-positive associations. The weighted median estimator was also used, which provides valid estimates when at least 50% of the information comes from valid instrumental variables46,47,48,49. Finally, the weighted mode-based estimation approach was employed, offering the most robust estimates when a majority of the instruments are invalid, yet still providing a consistent estimate of the true causal effect.
Mediation analysis
Two-step Mendelian randomization (MR), also referred to as network MR, is similar to the product-of-coefficients method. In two-step MR, estimates are derived by: (i) calculating the causal effect of the exposure on the mediator, and (ii) assessing the causal effect of the mediator on the outcome (Fig. S1C). In this study, we employed two-step MR for mediation based on the following evidence: (1) support for an effect of the trait on circulating immune cells and inflammatory proteins (step 1); (2) limited evidence for an effect of circulating immune cells and inflammatory proteins on the trait; and (3) evidence that circulating immune cells and inflammatory proteins influence the PD outcome of interest (step 2). A false discovery rate (FDR) of 5%, calculated using the Benjamini–Hochberg method, was applied as a threshold for determining supportive evidence of an association47. For the indirect effect of the mediator in the two-step MR mediation model, we multiplied the estimate for the effect of the trait on circulating immune cells and inflammatory protein levels (obtained from univariate MR) by the effect of circulating immune cells and inflammatory proteins on the PD outcome (also from univariate MR). The total effect was defined as the overall impact of the exposure on the outcome through all potential pathways, or as the effect of exposure on the outcome in the absence of any mediator (Fig. S1A).
Multivariable mendelian randomization (MVMR) analyses
MVMR regression estimates were compared to univariable MR estimates. For the univariable MR, we either used the same IVs as in the MVMR or employed a subset of IVs, which were retained after Steiger filtering between both the outcome and the exposure of interest, as well as between the exposure of interest and the other exposures. We report weak instrument bias via conditional F-statistics50 and heterogeneity through Cochran’s Q-statistic51.
Sensitivity analyses
In our analysis, only instrumental variables (IVs) that met the relevance, independence, and exclusion restriction assumptions were considered valid for the Mendelian randomization (MR) framework (Fig. S2). The validity of the first MR assumption was confirmed by establishing a significant association (p < 5 × 10⁻⁸) between all exposure IVs used in subsequent sensitivity analyses and the exposure. To verify the second assumption, we carefully selected independent IVs with a linkage disequilibrium (LD) value of less than 0.01 after clumping at a 1,000 kb range. Sensitivity analyses were then performed on these IVs that satisfied the first two MR assumptions. To assess the robustness of our findings, we employed a penalization method that reduces the influence of potentially problematic IVs—such as heterogeneous or outlier IVs—on the analysis50. Additionally, a systematic leave-one-out analysis was conducted to examine the potential for pleiotropy per SNP and its adherence to the exclusion restriction assumption. The impact of these analyses was evaluated using the penalized inverse-variance weighted (IVW) estimate. We also assessed the change in outcomes before and after the removal of each SNP. Heterogeneity among IVs was tested using Q statistics, with significance set at p ≤ 0.05. The strength of the instruments was evaluated by calculating the F-statistic51. To ensure adherence to the third MR assumption—the exclusion restriction assumption—we removed any instruments showing evidence of horizontal pleiotropy using multiple approaches, including PhenoScanner52. Horizontal pleiotropy was further examined using MR pleiotropy residual sum and outlier (MR-PRESSO) and MR-Egger regression, the latter based on its intercept terms53. Finally, a power analysis was conducted to determine the minimum effect size that could be reliably detected for each exposure54.
Statistical analysis
Multiple two-sample MR analyses were conducted to assess the causal effects of risk factors on PD-related outcomes. The IVW method was selected as the primary analysis for meta-analyzing individual Wald-type ratios of instrumental variables (IVs) using a random effects model, with odds ratios (ORs) reported per standard deviation increase in the risk factor level. The analyses utilized the TwoSampleMR (version 0.5.6), MR-cML (version 0.0.0.9), and MR-PRESSO (version 1.0) packages within R (version 3.4). The forest plots were created utilizing the ggplot2 package (version 2.0.1).
Results
Association of plasma phosphatidylcholine (17:0_18:1) levels with PD
Our first MR approach (IVW-MR) suggested that increased plasma phosphatidylcholine (17:0_18:1) levels are significantly associated with an increased risk of PD. In detail, our findings showed a substantial 24 percent increase in the risk of PD with each standard deviation (SD) rise in plasma phosphatidylcholine (17:0_18:1) levels (OR = 1.24; 95% confidence intervals (CI) = 1.07–1.45; p = 0.0046). Of note, this was in line with results from other robust MR analysis approaches (Fig. 1A). Despite the Simple mode MR testing for a causal relationship between plasma phosphatidylcholine (17:0_18:1) levels and the risk of PD being not significant (OR = 1.29; 95% confidence intervals (CI) = 0.89–1.86; p = 0.186), we found that the direction of Simple mode MR testing’s results is still in line with other approaches (Fig. 1A). Concurrently, we observed a potential causal link between PD risk and plasma triacylglycerol (51:4) and triacylglycerol (58:7) levels (Fig. 1B and C, and Supplementary table S1). More importantly, we also found a robust relationship between higher plasma phosphatidylcholine (17:0_18:1) levels and a higher risk of PD after further adjustment for triacylglycerol (51:4) and triacylglycerol (58:7) using multivariable Mendelian Randomization (MVMR) analysis. More specifically, there was a substantial 30% increase in the risk of PD with every SD rise in blood phosphatidylcholine (17:0_18:1) levels (OR = 1.30 for the IVW; p = 0.014; OR = 1.29 for the MR Egger, p = 0.023; OR = 1.30 for the Lasso, p = 0.015) (Fig. 1D).

Forest plot for the results from mendelian randomization (MR) analyses testing the effect of plasma lipidomes (including phosphatidylcholine (17:0_18:1), triacylglycerol (58:7), and triacylglycerol (51:4) levels) on the risk of PD.
The results of five MR analysis approaches were presented as odds ratios (ORs) and 95% confidence intervals (C.I.). P values less than 0.05 are indicated by *.
Association of plasma phosphatidylcholine (17:0_18:1) levels with circulating immune cells count
It was accepted that the metabolism of lipids is crucial in influencing inflammation in both short-term and long-term illnesses55,56. Thus, we examined the potential causative relationship between circulating immune cells and plasma phosphatidylcholine (17:0_18:1) levels. Interestingly, our MR analysis results indicated a causal relationship between plasma phosphatidylcholine (17:0_18:1) levels and the counts of 26 distinct immune cell subtypes. In detail, we identified a potential causal relationship between plasma phosphatidylcholine (17:0_18:1) levels and the counts of 13 distinct B lymphocyte subtypes. Among these 13 subtypes, only BAFF-R on CD20- B cells exhibited a negative causal association with plasma phosphatidylcholine (17:0_18:1) levels, where a 1 SD genetically instrumented increase in plasma phosphatidylcholine (17:0_18:1) levels was associated with a 13% reduction in BAFF-R on CD20- B cell count (95% CI -0.26 to -0.01; p = 0.034). In contrast, the counts of other B cell subsets were positively correlated with plasma phosphatidylcholine (17:0_18:1) levels. Specifically, a 1 SD increase in plasma phosphatidylcholine (17:0_18:1) levels was linked to an 18% increase in CD45 + B cell count (95% CI 0.048 to 0.30; p = 0.007), a 17% increase in CD27 + switched memory B cells (95% CI 0.05 to 0.29; p = 0.007), a 13% increase in CD27 + memory B cells (95% CI 0.01 to 0.25; p = 0.063), a 16% increase in CD27 + IgD- CD38dim B cells (95% CI 0.04 to 0.28; p = 0.011), a 17% increase in CD27 + IgD- CD38- B cells (95% CI 0.05 to 0.29; p = 0.007), a 16% increase in CD27 + CD24 + B cells (95% CI 0.01 to 0.26; p = 0.028), a 14% increase in CD27 + CD24 + B cells (95% CI 0.01 to 0.26; p = 0.028), a 16% increase in CD20 + switched memory B cells (95% CI 0.03 to 0.28; p = 0.014), a 13% increase in CD20 + memory B cells (95% CI 0.01 to 0.25; p = 0.043), an 18% increase in CD20 + IgD- CD38dim B cells (95% CI 0.06 to 0.30; p = 0.004), a 13% increase in CD20 + IgD- CD24- B cells (95% CI 0.01 to 0.26; p = 0.034), and a 13% increase in CD20 + CD24 + CD27 + B cells (95% CI 0.01 to 0.25; p = 0.041) (Fig. 2).

Forest plot for the results from mendelian randomization (MR) analyses testing the effect of plasma phosphatidylcholine (17:0_18:1) levels on counts of various subtypes’ circulating immune cells.
The results of five MR analysis approaches were presented as causal estimate and 95% confidence intervals (C.I.).
Additionally, we identified a potential causal relationship between increased plasma phosphatidylcholine (17:0_18:1) levels and elevated counts of six T cell subtypes. Specifically, a 1 SD genetically instrumented increase in plasma phosphatidylcholine (17:0_18:1) levels was associated with a 16% higher CD45 + lymphocyte count (95% CI 0.04 to 0.29; p = 0.014), a 14% higher CD45 + T cell count (95% CI 0.01 to 0.27; p = 0.037), a 13% higher CD45 + CD8 + T cell count (95% CI 0.01 to 0.25; p = 0.036), a 20% higher CD45 + HLA DR + T cell count (95% CI 0.07 to 0.33; p = 0.003), a 19% higher CD45 + HLA DR + CD4 + T cell count (95% CI 0.06 to 0.32; p = 0.005), and a 21% higher CD45 + HLA DR + CD8 + T cell count (95% CI 0.08 to 0.34; p = 0.002) (Fig. 2). Furthermore, our MR analysis indicated a potential causal relationship between plasma phosphatidylcholine (17:0_18:1) levels and the counts of myeloid cells, natural killer cells, monocytes, and granulocytes, as shown in Fig. 2.
Association of plasma phosphatidylcholine (17:0_18:1) levels with circulating inflammatory protein levels
In addition to investigating circulating immune cell subtypes, we also examined the causal association between plasma phosphatidylcholine (17:0_18:1) levels and circulating inflammatory proteins. Notably, we identified a negative causal relationship between increased plasma phosphatidylcholine (17:0_18:1) levels and seven circulating inflammatory protein levels. Specifically, a 1 SD genetically instrumented increase in plasma phosphatidylcholine (17:0_18:1) levels was associated with a 7% decrease in C-X-C motif chemokine 11 levels (95% CI − 0.14 to -0.01; p = 0.045), a 7% decrease in interleukin-17C levels (95% CI -0.15 to -0.01; p = 0.049), a 7% decrease in interleukin-2 receptor subunit beta levels (95% CI − 0.14 to − 0.01; p = 0.035), a 7% decrease in interleukin-8 levels (95% CI − 0.13 to − 0.01; p = 0.016), a 10% decrease in interleukin-7 levels (95% CI − 0.16 to -0.04; p = 0.000), a 7% decrease in monocyte chemoattractant protein 2 levels (95% CI − 0.14 to − 0.02; p = 0.013), and a 9% decrease in neurturin levels (95% CI − 0.17 to − 0.02; p = 0.018) (Fig. 3).

Forest plot for the findings of MR analyses testing the effect of plasma phosphatidylcholine (17:0_18:1) levels on levels of varied circulating proteins.
The results of five MR analysis methods were presented as the casual estimate and 95% confidence intervals (C.I.).
Association of circulating immune cells count with PD
Our MR analysis strongly supports the causal interpretation that the count of circulating immune cells is positively associated with the risk of PD. Specifically, increased counts of CD45 on CD14 + monocytes and CD45 on HLA DR + T cells were linked to an elevated risk of PD (IVW OR, 1.11 [95% CI 1.02–1.21], p = 0.021, and 1.06 [95% CI 1.01–1.13], p = 0.0475, respectively) (Fig. 4). This suggests that a 1 SD genetically instrumented increase in circulating CD45 on CD14 + monocytes and CD45 on HLA DR + T cells is associated with an 11% and 6% increase in PD risk, respectively. Notably, we found no evidence of a causal association between.

Forest plot for the findings of MR analyses testing the effect of circulating immune cells on the risk of PD.
circulating inflammatory protein levels and PD risk (Fig. 4).
The results of five MR analysis methods were presented as odds ratios (ORs) and 95% confidence intervals (C.I.). * indicates a p-value less than 0.05, ** denotes a p-value less than 0.01, and *** denotes a p-value less than 0.001.
Proportion of the association between plasma phosphatidylcholine (17:0_18:1) levels and PD mediated by circulating immune cells count
Notably, we did not observe a significant mediating effect in the association between plasma phosphatidylcholine (17:0_18:1) levels and the risk of PD. Specifically, the proportion of the effect mediated by circulating CD45 on CD14 + monocytes was 7% (p = 0.204), while the proportion mediated by circulating CD45 on HLA DR + T cells was 5.7% (p = 0.366) (Table 2). Interestingly, none of the 91 circulating inflammatory proteins, except for circulating immune cells, mediated the relationship between plasma phosphatidylcholine (17:0_18:1) levels and PD risk.
Sensitivity analysis
The results of our sensitivity analyses are presented in Tables S1 through S9 and include testing for pleiotropy and heterogeneity. Of note, we did not find any evidence of horizontal pleiotropy for the plasma phosphatidylcholine (17:0_18:1) levels, circulating immune cells, or circulating inflammatory proteins (p > 0.05). Also, there wasn’t evidence of heterogeneity for all analysis results (p > 0.05), which both displayed consistency between IVW and MR-Egger results.
Discussion
Genetic variant analyses have revealed that lipid metabolism and transport, along with autophagic-lysosomal and inflammatory pathways, represent significant genetic risk factors for age-related neurological disorders57,58,59. However, the precise molecular mechanisms by which these interactions contribute to the pathology of various neurodegenerative diseases, such as dementia, Alzheimer’s disease (AD), and PD, remain poorly understood. In this study, we identified a causal relationship between the plasma lipidome and PD, specifically causally linking elevated plasma phosphatidylcholine (17:0_18:1) levels to an increased risk of PD, as determined through Mendelian randomization (MR) analysis.
Lipid metabolism and PD risk have been the subject of several prior observational studies60,61,62. Still unknown, nevertheless, are the specific forms of disruption of lipid metabolism linked to PD risk. In the present study, we used MR analysis to examine the potential association between the risk of PD and the plasma lipidome, which contained 179 lipid species in 7174 Finnish adults. The MR findings demonstrated a causative relationship between alterations in plasma phosphatidylcholine (17:0_18:1) and the risk of PD. In agreement with our results, Smith J et al. discovered that there was a considerable up-regulation of two lipid species (phosphatidylcholine and lysophosphotidylcholine lipid classes) in the early stages of PD-like pathology27. The metabolite-based biomarker study by Maetzler W and colleagues also showed that the plasma of PD patients had higher levels of PC 44:6 and 44:563. Similarly, Öhman A et al. discovered that, in contrast to the control group, the atypical Parkinsonian disorders of the multiple system atrophy group had higher concentrations of the two phosphatidylcholine (PC) metabolites, PC ae C42:4 (fc = 1.21, p = 0.0256) and PC ae C44:5 (fc = 1.21, p = 0.0442)64. Remarkably, previous studies have also shown the opposite results: reduced levels of PC 34:2 and 46:2, PC 34:5, 36:5, and 38:5, as well as total PC, have been seen in the frontal cortex and plasma of PD patients65,66, as well as in the SN from PD patients who are solely male67. We suspect that the heterogeneity of PD patients and the variations in the metabolic alterations of various PC types in their blood may be the cause of this phenomenon. Furthermore, we inferred it may be due to confounding effects in observational studies, which MR analysis efficiently eliminates.
It is yet unknown how dysregulation of lipid metabolism, and particularly phosphatidylcholine (17:0_18:1), may lead to PD. Phosphatidylcholine is one of the most abundant phospholipids in cell membranes and is crucial for maintaining membrane integrity, cellular signaling, and fluidity68. It plays a central role in several cellular processes, including neurotransmission, and is involved in the formation of lipoproteins such as very-low-density lipoprotein (VLDL), which are critical for lipid transport and metabolism69. In the context of PD, there is growing interest in the role of lipid metabolism, particularly how alterations in lipid composition may influence the pathogenesis of neurodegenerative diseases70. Phosphatidylcholine, as part of membrane structures, may influence the stability and function of neuronal membranes, particularly in the central nervous system, which is critically involved in PD pathophysiology.
A major hallmark of PD is the aggregation of α-synuclein into toxic fibrils that form Lewy bodies in dopaminergic neurons. Studies have shown that lipid membranes, especially those enriched in phosphatidylcholine, are involved in the regulation of α-synuclein aggregation and propagation71. Elevated levels of phosphatidylcholine can enhance the membrane-binding of α-synuclein, which is essential for its aggregation. This occurs because α-synuclein interacts with phospholipids through its N-terminal lipid-binding region, and membrane lipids can influence the conformational transitions of α-synuclein, promoting its fibrillation72,73. Additionally, lipid compositions, including increased phosphatidylcholine levels, can alter the lipid bilayer’s microenvironment, which could facilitate α-synuclein’s conversion to a misfolded state prone to aggregation74. This increase in α-synuclein aggregation may further exacerbate neurotoxicity, leading to dopaminergic neuron degeneration.
Mitochondrial dysfunction is another key factor in PD pathogenesis. Phosphatidylcholine has been implicated in maintaining mitochondrial membrane integrity, and disturbances in phospholipid metabolism can impair mitochondrial function. Studies suggest that elevated phosphatidylcholine levels may alter the composition of mitochondrial membranes, leading to impaired oxidative phosphorylation, increased ROS production, and subsequent mitochondrial damage75. Increased ROS production can damage cellular components, including lipids, proteins, and DNA, ultimately leading to neuronal apoptosis and neurodegeneration76. Mitochondrial dysfunction also contributes to energy deficits in dopaminergic neurons, which are particularly vulnerable in PD. Therefore, this mitochondrial dysfunction is central to the pathogenesis of PD and may be exacerbated by alterations in lipid metabolism, including elevated phosphatidylcholine levels.
Lipid metabolism also plays a significant role in neuroinflammation, which is also a critical contributor to the progression of PD77. Phosphatidylcholine metabolism can influence the production of signaling molecules like lysophosphatidylcholine (LPC), which, when elevated, has been shown to activate inflammatory pathways78. LPC, generated by the hydrolysis of phosphatidylcholine, acts as a secondary messenger that can modulate the activity of immune cells, including microglia79. Microglia are the resident immune cells of the brain, and their activation is a hallmark of neuroinflammation in PD. Elevated LPC levels can induce the activation of microglia, which in turn release pro-inflammatory cytokines and reactive oxygen species (ROS), contributing to neuronal damage80. The inflammatory cascade initiated by activated microglia may exacerbate the neurodegenerative process in PD.
Lipid metabolism plays a pivotal role in the modulation of immune cell functions. Elevated phosphatidylcholine levels may influence circulating immune cells, such as monocytes, T cells, and microglia, which have been shown to contribute to neuroinflammation in PD81. Previous studies revealed a strong correlation between the onset and progression of PD and the activation of the NLRP3 inflammasome caused by endogenous danger signal molecules (such as monosodium urate, ATP, and amyloid-β)82,83,84. Recently, a growing body of evidence has demonstrated that endogenous oxidized phospholipids, like POVPC, have the capacity to modify the activation and polarization of phagocytes, including macrophages and dendritic cells, as well as to regulate cellular metabolism and pro-inflammatory signaling in these cells85,86. In addition, results from animal experiments indicated that endogenously generated oxidized phosphatidylcholines, including 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-phosphocholine (POVPC), activated the NLRP3 inflammasome, which caused macrophages to release IL-1β87. This implies that, at least in part, there is a connection between the onset of PD, the inflammatory response, and plasma lipid metabolites. Our results indicated that the causal relationship between elevated plasma phosphatidylcholine (17:0_18:1) levels and an increased risk of PD is partially mediated by immune cells. In line with these findings, Zhong et al. identified glycerophospholipid metabolism and arginine/proline metabolism as key altered metabolic pathways involved in systemic immune responses and low-grade inflammation88. Notably, Silvestre et al. found that dysregulated glycerophospholipid metabolism in Behçet’s disease contributes to a pro-inflammatory phenotype in circulating monocytes89. As a consequence, we believed that endogenous phosphatidylcholine that is oxidized under pathological conditions contributes to the progression of PD by regulating the inflammatory process.
Limitation
Understanding estimates derived from Mendelian randomization (MR) can be challenging due to the reliance on assumptions that may be difficult to test, such as the exclusion restriction, particularly when unmeasured or unknown confounders are present. One such potential confounder is horizontal pleiotropy, where genetic variants affect traits other than the outcome of interest. To mitigate the impact of horizontal pleiotropy on MR estimates, we employed sensitivity analyses that are less susceptible to this issue, specifically the MR-Egger regression and weighted median approaches. Furthermore, the GWAS data utilized in this study primarily includes individuals of European and East Asian descent, which may limit the generalizability of the findings to other populations. The lipid metabolism pathways and Parkinson’s disease (PD) risk factors could differ in populations with distinct genetic backgrounds, potentially affecting the observed associations. Finally, while the study suggests a potential mediating role of CD45 + CD14 + monocytes and CD45 + HLA-DR + T cells in the relationship between plasma phosphatidylcholine levels and PD risk, the mediation analysis results did not reach statistical significance. These findings warrant further investigation and validation in future studies to confirm their role in the pathophysiology of PD.
In conclusion, the present study found evidence supporting a potential causal relationship between higher risk of PD and higher plasma phosphatidylcholine (17:0_18:1) levels. And we also clarified a possible role for mediation through circulating immune cells (including CD45 on CD14 + monocytes and CD45 on HLA-DR + T), but not circulating inflammatory proteins. The molecular mechanisms underlying this association may involve pathways associated with inflammation or other mechanisms, but they could also operate independently of the classical regulatory mechanisms.
Data availability
The data and materials supporting the findings of this study are publicly available through the GWAS Catalog, repository [https://www.ebi.ac.uk/gwas/home] and references cited37,38,39, and40.
References
Van Den Eeden, S. K. et al. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 157(11), 1015–1022 (2003).
Lampropoulos, I. C., Malli, F., Sinani, O., Gourgoulianis, K. I. & Xiromerisiou, G. Worldwide trends in mortality related to Parkinson’s disease in the period of 1994–2019: Analysis of vital registration data from the WHO mortality database. Front. Neurol. 13, 956440 (2022).
Tysnes, O. B. & Storstein, A. Epidemiology of Parkinson’s disease. J Neural Transm. 124(8), 901–905 (2017).
Ou, Z. et al. Global trends in the incidence, prevalence, and years lived with disability of Parkinson’s disease in 204 countries/territories from 1990 to 2019. Front. Publ. Health. 9, 776847 (2021).
Dorsey, E. R. et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 17(11), 939–953 (2018).
Baizabal-Carvallo, J. F. & Jankovic, J. Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat. Rev. Neurol. 12(3), 175–185 (2016).
Schonhoff, A. M., Williams, G. P., Wallen, Z. D., Standaert, D. G. & Harms, A. S. Innate and adaptive immune responses in Parkinson’s disease. Prog. Brain Res. 252, 169–216 (2020).
Wallings, R. L., Humble, S. W., Ward, M. E. & Wade-Martins, R. Lysosomal dysfunction at the centre of Parkinson’s disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends Neurosci. 42(12), 899–912 (2019).
Juárez-Flores, D. L. et al. Disrupted mitochondrial and metabolic plasticity underlie comorbidity between age-related and degenerative disorders as Parkinson disease and type 2 diabetes mellitus. Antioxidants 9(11), 1063 (2020).
Robea, M. A. et al. Parkinson’s disease-induced zebrafish models: Focussing on oxidative stress implications and sleep processes. Oxid. Med. Cell Longev. 2020, 1370837 (2020).
Raza, C., Anjum, R. & Shakeel, N. U. A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 226, 77–90 (2019).
Grayson, M. Parkinson’s disease. Nature 538(7626), S1 (2016).
Mori, A., Imai, Y. & Hattori, N. Lipids: Key players that modulate α-synuclein toxicity and neurodegeneration in Parkinson’s disease. Int. J. Mol. Sci. 21(9), 3301 (2020).
Neumann, J. et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132(Pt 7), 1783–1794 (2009).
Sidransky, E. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361(17), 1651–1661 (2009).
Gan-Or, Z. et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 80(17), 1606–1610 (2013).
Chang, D. et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49(10), 1511–1516 (2017).
Robak, L. A. et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140(12), 3191–3203 (2017).
Simón-Sánchez, J. et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 41(12), 1308–1312 (2009).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46(9), 989–993 (2014).
Goldschmidt, H. L. et al. DGKθ catalytic activity is required for efficient recycling of presynaptic vesicles at excitatory synapses. Cell Rep. 14(2), 200–207 (2016).
Puchkov, D. & Haucke, V. Greasing the synaptic vesicle cycle by membrane lipids. Trends Cell Biol. 23(10), 493–503 (2013).
Do, C. B. et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 7(6), e1002141 (2011).
Lee, J. H. et al. SREBP-1a-stimulated lipid synthesis is required for macrophage phagocytosis downstream of TLR4-directed mTORC1. Proc. Natl. Acad. Sci. USA. 115(52), E12228–E12234 (2018).
Shahmoradian, S. H. et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22(7), 1099–1109 (2019).
Klein, J. Membrane breakdown in acute and chronic neurodegeneration: Focus on choline-containing phospholipids. J Neural Transm. 107(8–9), 1027–1063 (2000).
Farmer, K., Smith, C. A., Hayley, S. & Smith, J. Major alterations of phosphatidylcholine and lysophosphotidylcholine lipids in the substantia Nigra using an early-stage model of Parkinson’s disease. Int. J. Mol. Sci. 16(8), 18865–18877 (2015).
Shamim, A., Mahmood, T., Ahsan, F., Kumar, A. & Bagga, P. Lipids: An insight into the neurodegenerative disorders. Clin. Nutr. Exp. 20, 1–19 (2018).
Wright, M. M., Howe, A. G. & Zaremberg, V. Cell membranes and apoptosis: Role of cardiolipin, phosphatidylcholine, and anticancer lipid analogues. Biochem. Cell Biol. 82(1), 18–26 (2004).
Macphee, C. H. Lipoprotein-associated phospholipase A2: a potential new risk factor for coronary artery disease and a therapeutic target. Curr. Opin. Pharmacol. 1(2), 121–125 (2001).
Taylor, L. A., Arends, J., Hodina, A. K., Unger, C. & Massing, U. Plasma lyso-phosphatidylcholine concentration is decreased in cancer patients with weight loss and activated inflammatory status. Lipids Health Dis. 6, 17 (2007).
Goonesinghe, A. et al. Pro-apoptotic Bid induces membrane perturbation by inserting selected lysolipids into the bilayer. Biochem. J. 387(Pt 1), 109–118 (2005).
Yun, D. H., Jeon, E. S., Sung, S. M., Ryu, S. H. & Kim, J. H. Lysophosphatidylcholine suppresses apoptosis and induces neurite outgrowth in PC12 cells through activation of phospholipase D2. Exp. Mol. Med. 38(4), 375–384 (2006).
Mulder, C. et al. Decreased lysophosphatidylcholine/phosphatidylcho—line ratio in cerebrospinal fluid in Alzheimer’s disease. J. Neural. Transm. (Vienna). 110(8), 949–955 (2003).
Drzazga, A., Sowińska, A. & Koziołkiewicz, M. Lysophosphatidylcholine and lysophosphatidylinosiol-novel promissing signaling molecules and their possible therapeutic activity. Acta. Pol. Pharm. 71(6), 887–899 (2014).
Fuchs, B., Schiller, J., Wagner, U., Häntzschel, H. & Arnold, K. The phosphatidylcholine/lysophosphatidylcholine ratio in human plasma is an indicator of the severity of rheumatoid arthritis: Investigations by 31P NMR and MALDI-TOF MS. Clin. Biochem. 38(10), 925–933 (2005).
Ottensmann, L. et al. Genome-wide association analysis of plasma lipidome identifies 495 genetic associations. Nat. Commun. 14(1), 6934 (2023).
Orrù, V. et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat. Genet. 52(10), 1036–1045 (2020).
Zhao, J. H. et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat. Immunol. 24(9), 1540–1551 (2023).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53(10), 1415–1424 (2021).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
van der Graaf, A. et al. Systematic prioritization of candidate genes in disease loci identifies TRAFD1 as a master regulator of IFNγ signaling in celiac disease. Front Genet. 11, 562434 (2020).
Hemani, G. et al. The MR-base platform supports systematic causal inference across the human phenome. Elife 30(7), e34408 (2018).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 40(4), 304–314 (2016).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44(2), 512–525 (2015).
Slob, E. A. W. & Burgess, S. A comparison of robust Mendelian randomization methods using summary data. Genet. Epidemiol. 44(4), 313–329 (2020).
Davey Smith, G. & Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23(R1), R89-98 (2014).
Sanderson, E., Spiller, W. & Bowden, J. Testing and correcting for weak and pleiotropic instruments in two-sample multivariable Mendelian randomization. Stat. Med. 40(25), 5434–5452 (2021).
Bowden, J., Hemani, G. & Davey, S. G. Invited commentary: Detecting individual and global horizontal pleiotropy in Mendelian randomization-a job for the humble heterogeneity statistic?. Am. J. Epidemiol. 187(12), 2681–2685 (2018).
Burgess, S., Bowden, J., Fall, T., Ingelsson, E. & Thompson, S. G. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology 28(1), 30–42 (2017).
Pierce, B. L., Ahsan, H. & Vanderweele, T. J. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int. J. Epidemiol. 40(3), 740–752 (2011).
Staley, J. R. et al. PhenoScanner: A database of human genotype-phenotype associations. Bioinformatics 32(20), 3207–3209 (2016).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50(5), 693–698 (2018).
Brion, M. J., Shakhbazov, K. & Visscher, P. M. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiol. 42(5), 1497–1501 (2013).
van Diepen, J. A., Berbée, J. F., Havekes, L. M. & Rensen, P. C. Interactions between inflammation and lipid metabolism: Relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis 228(2), 306–315 (2013).
Bennett, M. & Gilroy, D. W. Lipid mediators in inflammation. Microbiol Spectr. https://doi.org/10.1128/microbiolspec.MCHD-0035-2016 (2016).
Shi, H. et al. Genetic variants influencing human aging from late-onset Alzheimer’s disease (LOAD) genome-wide association studies (GWAS) Neurobiol Aging. Neurobiol. Aging. 33(8), 1849-e5 (2012).
Klemann, C. et al. Integrated molecular landscape of Parkinson’s disease. NPJ Parkinsons Dis. 3, 14 (2017).
Chang, D. et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516 (2017).
Wang, Q., Liu, Y. & Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 4, 19 (2015).
Kubo, S. et al. A combinatorial code for the interaction of alpha-synuclein with membranes. J. Biol. Chem. 280, 31664–31672 (2005).
Coulombe, K. et al. Impact of DHA intake in a mouse model of synucleinopathy. Exp. Neurol. 301, 39–49 (2018).
Stoessel D, Schulte C, Teixeira Dos Santos MC, Scheller D, Rebollo-Mesa I, Deuschle C, Walther D, Schauer N, Berg D, Nogueira da Costa A, Maetzler W. Promising metabolite profiles in the plasma and CSF of early clinical Parkinson’s disease. Front. Aging Neurosci. 2018; 10: 51.
Pathan, M., Wu, J., Lakso, H. Å., Forsgren, L. & Öhman, A. Plasma metabolite markers of Parkinson’s disease and atypical Parkinsonism. Metabolites 11(12), 860 (2021).
Zhang, J., Zhang, X., Wang, L. & Yang, C. High performance liquid chromatography-mass spectrometry (LC-MS) based quantitative lipidomics study of ganglioside-NANA-3 plasma to establish its association with Parkinson’s disease patients. Med Sci Monit. 23, 5345–5353 (2017).
Wood, P. L., Tippireddy, S., Feriante, J. & Woltjer, R. L. Augmented frontal cortex diacylglycerol levels in Parkinson’s disease and Lewy body disease. PLoS ONE 13(3), e0191815 (2018).
Seyfried, T. N. et al. Sex-related abnormalities in substantia Nigra lipids in Parkinson’s disease. ASN Neuro 10, 1759091418781889 (2018).
Akhberdi, O. et al. Roles of phospholipid methyltransferases in pycnidia development, stress tolerance and secondary metabolism in the taxol-producing fungus Pestalotiopsis microspore. Microbiol. Res. 210, 33–42 (2018).
Wurtman, R. J. How anticholinergic drugs might promote Alzheimer’s disease: More amyloid-β and less phosphatidylcholine. J. Alzheimers Dis. 46(4), 983–987 (2015).
Fabelo, N. et al. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 17(9–10), 1107–1118 (2011).
Zhaliazka, K., Ali, A. & Kurouski, D. Phospholipids and cholesterol determine molecular mechanisms of cytotoxicity of α-synuclein oligomers and fibrils. ACS Chem. Neurosci. 15(2), 371–381 (2024).
Trostchansky, A. et al. Interaction with phospholipids modulates alpha-synuclein nitration and lipid-protein adduct formation. Biochem. J. 393(Pt 1), 343–349 (2006).
Dou, T. & Kurouski, D. Phosphatidylcholine and phosphatidylserine uniquely modify the secondary structure of α-synuclein oligomers formed in their presence at the early stages of protein aggregation. ACS Chem. Neurosci. 13(16), 2380–2385 (2022).
Musteikytė, G. et al. Interactions of α-synuclein oligomers with lipid membranes. Biochim. Biophys. Acta Biomembr. 1863(4), 183536 (2021).
Shiino, H. et al. Chemical inhibition of phosphatidylcholine biogenesis reveals its role in mitochondrial division. iScience. 27(3), 109189 (2024).
Sesti, F., Liu, S. & Cai, S. Q. Oxidation of potassium channels by ROS: a general mechanism of aging and neurodegeneration?. Trends Cell Biol. 20(1), 45–51 (2010).
Andersen, C. J. Lipid metabolism in inflammation and immune function. Nutrients 14(7), 1414 (2022).
Aiyar, N. et al. Lysophosphatidylcholine induces inflammatory activation of human coronary artery smooth muscle cells. Mol. Cell Biochem. 295(1–2), 113–120 (2007).
Ren, J., Lin, J., Yu, L. & Yan, M. Lysophosphatidylcholine: potential target for the treatment of chronic pain. Int. J. Mol. Sci. 23(15), 8274 (2022).
Xie, Y. et al. Transforming growth factor-β1 protects against LPC-induced cognitive deficit by attenuating pyroptosis of microglia via NF-κB/ERK1/2 pathways. J Neuroinflammation. 19(1), 194 (2022).
Hughes, H. K., Moreno, R. J. & Ashwood, P. Innate immune dysfunction and neuroinflammation in autism spectrum disorder (ASD). Brain, Behav. Immun. 1(108), 245–54 (2023).
Caseley, E. A., Poulter, J. A. & Rodrigues, F. Immunome project consortium for autoinflammatory disorders (ImmunAID) Caseley Emily A. 1 Poulter James A. 1, McDermott MF. Inflammasome inhibition under physiological and pharmacological conditions. Genes Immun. 21(4), 211–23 (2020).
Han, X. et al. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for Parkinson disease. Autophagy 15(11), 1860–1881 (2019).
Ahmed, S., Kwatra, M., Ranjan Panda, S., Murty, U. S. N. & Naidu, V. G. M. Andrographolide suppresses NLRP3 inflammasome activation in microglia through induction of parkin-mediated mitophagy in in-vitro and in-vivo models of Parkinson disease. Brain Behav Immun. 91, 142–158 (2021).
Di Gioia, M. & Zanoni, I. Dooming phagocyte responses: Inflammatory effects of endogenous oxidized phospholipids. Front Endocrinol. 15(12), 626842 (2021).
Qin, J., Goswami, R., Balabanov, R. & Dawson, G. Oxidized phosphatidylcholine is a marker for neuroinflammation in multiple sclerosis brain. J. Neurosci. Res. 85(5), 977–984 (2007).
Yeon, S. H., Yang, G., Lee, H. E. & Lee, J. Y. Oxidized phosphatidylcholine induces the activation of NLRP3 inflammasome in macrophages. J. Leukoc. Biol. 101(1), 205–215 (2017).
Zhu, Q. et al. Comprehensive metabolic profiling of inflammation indicated key roles of glycerophospholipid and arginine metabolism in coronary artery disease. Front. Immunol. 8(13), 829425 (2022).
Mendes-Frias, A. et al. Dysregulation of glycerophospholipid metabolism during Behçet’s disease contributes to a pro-inflammatory phenotype of circulating monocytes. J. Transl. Autoimmun. 11(3), 100056 (2020).
Funding
This research was generously supported by the National Natural Science Foundation of China (Project No. 821713273), National Clinical Key Specialty (Neurosurgery) Project of China, and the High level medical and health talents in Jinan City, and Shandong Province Medical and Health Science and Technology Development Program Project (Project No. 202204040490).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Y.J.J. and H.L.Z. contributed to the conceptualization. Y.M.C., Q.H., and D.Q.S. contributed to the literature search. Y.J.J. and J.Y.S. contributed to the data analysis. Y.J.J., J.Y.S., and D.Q.S. contributed to the writing of the original draft. D.Z.K., L.Y.H., and D.L.W. contributed to the writing, review, and editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study only involves publicly available GWAS statistical abstracts and does not capture individual-level data, therefore is not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, D., Jing, Y., Su, J. et al. Higher levels of plasma phosphatidylcholine (17:0_18:1) raise the risk of developing Parkinson’s disease. Sci Rep 15, 28093 (2025). https://doi.org/10.1038/s41598-025-12054-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12054-y
