
Abstract
Anorexia nervosa (AN) is a mental disorder marked by a significantly low body weight. Differentially methylated CpG sites have been reported to be involved in body weight regulation. Methylation pattern may change during considerable weight gain by in-patient treatment. Consequently, we aimed to (1) replicate the hypomethylation at the NR1H3 gene locus (identified in our previous epigenome-wide association study) in independent study groups of 189 female patients with AN and 67 healthy-lean female controls, and (2) identify regions associated with large weight gain associated DNA methylation changes in three patients with AN through whole-genome bisulfite sequencing in CD14+ cells. In the replication study, no evidence was observed for hypomethylation at the investigated 15 CpG sites of the NR1H3 locus. Relying on two analysis tools (camel, metilene) to identify differentially methylated regions (DMRs), subtle methylation differences concordant between both tools were detected only when the usual threshold of camel was lowered. Then, eight regions were selected exemplarily for technical replication with deep bisulfite sequencing in the same three patients with AN. None of the regions could be confirmed. Summarising, we could not confirm hypomethylation at NR1H3 and could not detect methylation differences in patients with AN between admission and weight gain at discharge.
Similar content being viewed by others
Introduction
Human body weight is regulated by both genetic and environmental factors1. Anorexia nervosa (AN) is a severe, potentially life-threatening mental disorder that typically manifests during late childhood and adolescence2,3. Females are approximately ten times more often diagnosed than males with lifetime prevalence rates of 0.8% in females in the US2. The diagnosis is based on a significantly low body weight and AN-related psychopathology including weight and shape concerns4. Starvation is a core physiological feature of actue AN5. Accordingly, the symptomatology results from an age-, duration- and severity-dependent intertwining of primary cognitive and behavioral features with starvation induced somatic, cognitive and behavioral symptoms5.
Previously, it has been shown, that caloric restriction alters DNA and histone modifications (acetylation, methylation)6,7. For instance, it was shown that individuals who were prenatally exposed to famine (Dutch Hunger Winter in 1944–45) showed six decades later reduced DNA methylation at the imprinted IGF2 gene locus, when compared with unexposed, same-sex siblings8.
For AN, epigenetic studies are still scarce9,10. So far, the DNA methylation of a number of candidate genes has been analysed (e.g.11,12,13,14,15,16,17,18,19) and solely few epigenome-wide association studies (EWAS) have been performed20,21,22,23,24 (reviewed by 9,10). Given that DNA methylation patterns are highly cell-type specific25,26, it remains uncertain whether the previously reported DNA methylation changes indeed reflect true alterations due to diseases and/or starvation, respective interventions or whether the detected changes merely reflect differences in the cellular composition of the investigated tissue. All previously published studies investigated methylation patterns in whole blood, DNA from saliva or buccal cells10. Moreover, factors like infections27, menstrual cycle28 and stress29 influence the highly dynamic cell composition of leukocytes. Thus, sampling at different time points might induce DNA methylation differences30.
Previously, we examined DNA methylation derived from whole blood of 47 females with AN, as well as two control groups of 47 lean females without AN and 100 population-based females21. We applied two reference-free methods (FastLMM-EWASher, RefFreeEWAS) to account for different cell type compositions and searched for consensus CpG sites identified by both methods. In this study, 51 consensus sites were identified in AN versus lean and 81 in AN versus population-based comparisons. These sites had not previously been reported in AN methylation analyses. By comparing the consensus sites with the literature and a genome-wide association study meta-analysis, a single nucleotide polymorphism (rs923768) in CSGALNACT1 was identified to be nominally associated with AN, while at gene level, hypermethylation at TNXB in patients with AN compared to controls was confirmed21. Our analysis of AN versus lean controls further revealed evidence for a relevant locus at NR1H321. However, the methylation was opposite to the one previously reported20. Collectively, we confirmed genes like TNXB described to comprise differentially methylated sites, and highlighted further sites that might be specifically involved in AN21.
In accordance, Steiger and colleagues conducted a genome-wide DNA methylation analysis in whole blood of 75 female patients with AN, 31 females with remitted AN and 41 females with no diagnosed eating disorder. Hereby, methylation levels and directions differed between individuals suffering from acute AN and patients with remitted AN indicating reversible epigenetic alterations22. Likewise, a larger study demonstrated reversible methylation patterns in individuals with AN23. An investigation of whole blood samples of seven monozygotic AN-discordant twin pairs revealed nine gene-linked differentially methylated CpG sites. When validating these with a rank regression model as well as a beta regression, two and six CpG sites associated with genes remained significant, respectively. The correlated genes were mainly linked to metabolic and psychological traits31.
Within the present study, we aim to investigate methylation changes in patients with AN in more detail. First, as we identified a hypomethylation at NR1H3 within our EWAS approach21 in contrast to the previously reported hypermethylation20,23, we aimed to replicate the hypomethylation at NR1H3 in 198 female patients with AN and 87 healthy-lean female controls using whole blood samples. Secondly, we aim to validate the hypothesis that a weight gain achieved in in-patient treatment considerably alters the methylome across a wide range of cell and tissue types. Therefore and to further address the above-mentioned limitations of DNA methylation studies, we aimed to identify de novo and genome-wide differentially methylated regions (DMRs) before and after weight gain in three patients with AN by performing whole-genome bisulfite sequencing (WGBS) of DNA isolated from the CD14+ cell population. As patients with AN tend to gain a considerable amount of weight during short-term interventions, we assume that if a link between DNA methylation and body weight regulation exists, we should be able to observe this link when analysing these patients. Subsequently, we conducted a technical replication of the detected de novo DMRs with deep bisulfite sequencing in these same three patients. This study design can reduce the amount of false positives resulting from variation in DNA sequence and cell type composition, which are the major confounding factors in DNA methylation studies32.
Results
Replication analyses of the NR1H3 gene locus
In the independent study subgroup, we identified at six CpG sites nominal differences in the mean methylation values between females with AN (n = 189) and healthy-lean controls (n = 67; see Table 1; CpG sites #1, #2, #10, #11, #13 and #14). Among them, two showed a hypomethylation in females with AN (#1 and #2), while four were hypermethylated (#10, #11, #13 and #14). Yet, the methylation differences were small (see Table 1). These results are consistent with those from the dependent study subgroup, i.e. the complete sample. One of the identified CpG sites (#11) was also included in our previous study21 that accounted for the underlying cell type distributions33,34. There, no evidence for methylation differences could be observed for #11. Note, in the previous study, we observed the hypomethylation at NR1H3 on the gene level, but not at CpG site level.
Identification of putatively weight gain associated DMRs
Within our exploratory study in three female patients with AN receiving in-patient treatment, in which all females gained considerably weight (see Table 2), we did not observe evidence for DMRs between admission and weight gain based on camel applying the proposed thresholds. If we lowered the thresholds and increased the stringency, we identified 35 DMRs (see Supplementary Table 4). Relying on metilene, we observed 30 DMRs (see Supplementary Table 5). Thirteen DMRs were detected with both tools (see Supplemenatry Tables 4 and 5). With regard to our DMR identification and based on the publication by Ziller et al.35, we estimated that we reached a true positive rate of about 80% and a false positive rate of about 20%.
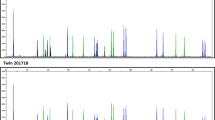
The principal component analysis (PCA) did not reveal a stringent grouping of the samples according to pre- and post-intervention (see Supplementary Fig. 1). Similarly, when performing a cluster analysis of the 1,000 most variable CpG sites, each individual showed distinct methylation patterns (see Fig. 1). These detected inter-individual methylation differences most likely reflect the different genetic backgrounds.

Cluster analysis of the 1,000 most variable CpG sites. The obtained methylomes of pre- (T0) and post-treatment-induced weight gain (T1) from three female patient with anorexia nervosa (AN) are shown.
Technical replication of WGBS-resultant targets
Based on the DMR identification (using the decreased threshold), we aimed to technically replicate eight regions (four detected by both DMR calling tools, and four detected by camel; see Supplementary Tables 4 and 5) using deep bisulfite sequencing (DBS) in the same three individuals (pre- and post- weight gain, see Table 2). These regions comprised: DMR-1 within Galactosidase Beta 1 Like (GLB1L), intergenic DMR-3, DMR-7 within Family With Sequence Similarity 50 Member B (FAM50B), DMR-11 within Mesoderm Specific Transcript (MEST), DMR-13 within ER Lipid Raft Associated 2 (ERLIN2), DMR-16 within Exonuclease 3’-5’ Domain Containing 3 (EXD3), DMR-22 within SNRPN Upstream Reading Frame/Small Nuclear Ribonucleoprotein Polypeptide N (SNURF/SNRPN) and DMR-28 within Heat Shock Protein Family A (Hsp70) Member 12B (HSPA12B; see Supplementary Table 4). All CpG sites contained in the DMRs were included in the amplicons of the corresponding polymerase chain reactions (PCRs; see Supplementary Table 1). None of the methylation differences identified by WGBS could be confirmed in any of the investigated regions (see Fig. 2).

Technical replication of eight WGBS-derived target regions from three female patient with anorexia nervosa (AN) for pre- (T0) and post-treatment induced weight gain (T1) using DBS. DBS, deep bisulfite sequencing; WGBS, whole-genome bisulfite sequencing.
Discussion
Evidence for effects of nutrition on the methylome have emerged (e.g.6,7,8). Yet, the impact of DNA methylation on AN and related symptoms like starvation remain ambiguous10. We have previously performed a high-throughput DNA methylation analysis (EWAS) and detected some CpG sites differentially methylated in AN with one locus at NR1H321 exhibiting deviating methylation directions compared to an earlier study20. Consequently, we aimed to replicate our previous findings21. In our replication approach for NR1H3, despite a larger sample size, we did not observe evidence for methylation differences between patients with AN and healthy-lean female controls at the analysed CpG sites at NR1H3. This contrasts with previous reports from other groups describing a hypermethylation at this site20,23. Unlike our former study, we were unable to correct for cell type compositions, as EWAS data were not available for all participants. Without this correction, there is also no evidence for deviations in the data sets of the previous, here re-analysed EWAS study21. Summarising, we were unable to replicate methylation differences at NR1H3, regardless of the methylation direction previously reported20,21.
Subsequently, we analysed weight gain associated DMRs in three patients with AN receiving in-patient treatment. We assumed that, if rapid increases in body weight (within weeks) are associated with methylation changes, the effect of a significant weight gain between admission and discharge on methylation changes should be large and, consequently, detectable in such patients with AN. By comparing DNA methylation patterns in the same individuals pre- and post-weight gain, differences based on genetic variation can be ruled out. However, no evidence for methylation differences were observed, and DMRs detected at a lower than standard thresholds could not be technically confirmed. Generally, our study is underpowered to detect subtle methylation differences, such as methylation changes present in only a few CD14+ cells. However, we aimed to detect large differences as previously suggested36. While we were unable to confirm our hypothesis that the pronounced weight gain during in-patient treatment heavily alters the DNA methylation, it remains possible that such changes may only become evident after a longer period of time.
Other studies trying to replicate previous findings revealed similar outcomes. For example, one study failed to replicate18 the previously reported overall global hypermethylation found on a CpG site at HDAC420.Yet, this replication attempt might be hampered by the use of different initial source tissues18. In a different study31, genome-wide methylation of monozygotic twins discordant for AN were analysed and nine CpG sites at genes were detected. Subsequently, the authors were able to validate their results at least partially in non-familial AN samples and controls applying rank regression and beta regression modelling (two and six prior detected CpG sites, respectively).
In general, many studies have investigated candidate-gene approaches (e.g.11,12,13,14,15,16,17,18,19) and some investigated genome-wide methylation pattern (e.g.20,21,31). However, it remains uncertain whether the detected methylation changes and differences are due to AN, its related starvation or due to a varying cell type composition. This is particularly evident, as it is known that cell type composition varies between controls and patients with AN as well as between patients with AN before and after treatment37. For instance, it was found that patients with AN had reduced levels of lymphocytes and B-cells after discharge following a six-week treatment in comparison to healthy individuals37. Of note, blood is not a primary tissue for AN. Yet, it is easily accessible, can be obtained with minimal invasiveness during routine clinical procedures and allows for sampling of large number of individuals. In fact, primary tissues such as brain or adipose tissue are not ethically feasible to obtain from patients and particularly from controls. In our weight gain-associated DMR identification approach, we decided to use a homogenous cell type (CD14+ cells). Nevertheless, different results might be obtained if different cell types would have been chosen (e.g. T-cells, NK-cells, etc.) as DNA methylation is highly cell type-specific25,26. Further, we have exclusively analysed three patients with AN. DNA methylation might be highly variable between individuals and thus more subjects would be needed to detect subtle common effects.
It is remarkable that we could not observe evidence for an impact on the methylome by the pronounced weight gain during in-patient treatment. This might be due to the fact that cell type-specific methylation patterns are highly stable. This stability can probably be attributed to stem or progenitor cells. We previously showed differences in metabolic profiles in patients with AN compared to controls and in patients with AN between acute stage of starvation and after a short-term weight recovery. A substantial number of metabolite levels did not return to normal after short term recovery38. Yet, after a prolonged period of time, the metabolite levels resembled those of healthy controls39. Hence, one might hypothesise that weight gain could exert an effect on the methylome, albeit at a later time point. Putatively, additional factors like the nutritional status24 might also be implicated in methylation changes.
Differences in demographic characteristics such as age, weight and BMI were observed between the included study groups (see Table 3). However, these differences are inherently linked to the clinical phenotype of AN, which is typically characterized by markedly reduced body weight and a higher prevalence among adolescent females2,3,4. As such, strict matching on these parameters may not be clinically representative and could obscure relevant disease-specific features. Yet, age is a well-established modulator of DNA methylation, with promoter-associated CpG sites often exhibiting hypermethylation and other genomic regions showing hypomethylation over time40,41,42,43. Longitudinal data reported a median rate of methylation change of approximately 0.18% per year44, indicating rather subtle age-related effects. Notably, the inter-individual variability in methylation patterns can be of similar magnitude to age-related changes44,45. Additionally, lifestyle factors such as physical activity, diet and stress have been shown to influence DNA methylation itself as well as the pace of epigenetic aging42,45,46. Even among monozygotic twins, DNA methylation patterns become increasingly divergent over time, particularly when exposed to distinct environmental influences43. Thus, epigenetic aging and dynamics are governed by a complex interplay of various determinants. While we cannot entirely exclude the possibility that differences in age, weight and BMI may introduce a certain degree of confounding, these variables represent only a subset of the full spectrum of factors influencing the epigenome. In fact, they are closely linked to the clinical manifestation of AN and should thus be considered group-specific characteristics.
In sum, we could not replicate findings of our previous EWAS approach21 and studies by others20. Furthermore, we were unable to detect weight gain associated methylation patterns.
Materials and methods
Samples: definition and characteristics
Replication study group: The data set for the replication analyses of the NR1H3 gene locus comprised 285 females in total (see Table 3). Among them, 198 females (69.47%) were previously diagnosed with AN, while 87 healthy-lean females (30.53%) had no diagnosis of AN or any other eating disorder and thus acted as controls. All participants were recruited in Germany. Of the included participants, 29 females (9 with AN, 20 without AN) were already analysed in the high-throughput DNA methylation analyses21. Consequently, we additionally defined the subgroup of females that were not included in the previous work (“independent subgroup”) and of females included in the previous work (“non-independent subgroup”). To compare selected patient characteristics, we applied the non-parametric Mann–Whitney U test (using IBM SPSS Statistics 29.0.0 for Windows) and reported the two-sided p-values. The phenotypic characteristics of the study groups are depicted in Table 3. Patients with AN and controls differed in all reported parameters.
DMR identification cohort: For the genome-wide identification of DMRs, three female patients with AN receiving in-patient treatment at the Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy at the University Hospital Essen were included. Inclusion criteria were diagnosis of AN according to DSM-IV-TR47, female sex and German ancestry (characteristics: see Table 2). AN diagnosis was confirmed via a clinical examination and a semi-structured interview (Kiddie Schedule for Affective Disorders and Schizophrenia; K-SADS)48. Adolescent patients with AN participated in a multimodal multidisciplinary treatment program49 based on dialectical behavioural therapy for adolescents that included weight restoration, individual nutritional counselling, individual therapy (twice a week), skill-groups, individual skill training (twice a week), family sessions (fortnightly), and a group psychoeducation program for parents (monthly). During weight restoration, patients received nutritional counselling and meal support training. Therapy goals were a weight gain of at least 500 g per week and a minimum target weight in the range of the 10-20th age-adjusted BMI percentile.
The complete study was approved by the local ethics committee of the Faculty of Medicine at the University Hospital Essen (No. 06-3212 amendment of November 2016). Patients and in case of minors their parents gave written informed consent. This study was conducted in accordance to the Declaration of Helsinki50.
Replication analyses of the NR1H3 gene locus
For the statistical evaluation of the replication, we used two analysis sets. The main analysis was performed only with females not included in the original work21 to ensure an independent replication. A sensitivity analysis was conducted in the complete sample, i.e. including females from both the independent and the non-independent subgroup. These two analysis sets are subsequently called “independent sample” (containing only females from the independent subgroup) and “non-independent sample” (complete sample).
Within the replication study group, blood was drawn from each participant and DNA was isolated from whole blood using a salting out approach51. DNA was bisulfite-modified using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s instructions. To eventually allow a pooled sequencing approach, a two-step PCR (Veriti 96-well Thermal Cycler, Applied Biosystems, Foster City, CA, USA) was conducted. The first PCR was performed using NR1H3-specific primers with tags at the 5’ end (see Supplementary Table 1). Subsequently, the PCR products were used in a second PCR applying primers with forward and reverse adapters complementary to the tags attached in the first PCR round (see Supplementary Table 2). The usage of unique combinations of forward and reverse adapters allowed an unambiguous assignment of each sample despite a pooled sequencing. PCR products of the second PCR were subsequently purified using the QIAquick 96 PCR Purification Kit (QIAGEN, Venlo, Netherlands). Library preparation and sequencing on the HiSeq2500 Illumina sequencer was performed at the BioChip Lab of the Center of Medical Biotechnology (ZMB) of the University of Duisburg-Essen. Raw sequencing reads were analysed using the amplikyzer252 which enables demultiplexing of the data, creation of FASTQ files and provides the output of DNA methylation values in percent for each CpG.
To investigate the association between the female’s AN status (with AN, healthy-lean controls without AN) and DNA methylation, a robust linear regression modelling was applied for each CpG site. Each model comprised the site-specific DNA methylation value as a dependent variable and the indicator for AN diagnosis (1: AN; 0: no AN) as an independent variable. Due to the candidate CpG site approach in this study, an adjustment for different cell type compositions was not possible. For the robust linear model, MM estimation was applied and the Tukey bi-square function (with the tuning constant c = 4.685) for observation weighting was used. We provide the estimated DNA methylation difference (regression coefficient) together with the 95% confidence interval. A two-sided significance level of 0.05 was used and was not adjusted for multiple comparisons. Analyses were performed with R (version 3.5.1) and the R package MASS (v.7.3–51.4).
Identification of weight gain associated DMRs
In the DMR identification cohort, whole blood was drawn at admission and discharge. Given the cell type specificity of DNA methylation25,26,33, this analysis focused on one homogenous cell population, namely the CD14+ monocytes. Monocytes have been demonstrated to play a pivotal role in inflammatory responses53, and patients with AN who are acutely ill exhibit a dysregulated inflammatory status54. Furthermore, CD14+ monocytes have been utilised in epigenetic studies of comorbid psychiatric disorders, such as major depressive disorder55, and metabolic conditions, such as the metabolic syndrome56, which constitute the opposite end of the BMI spectrum. Therefore, patients’ blood was used to immunomagnetically purify DNA of CD14+ monocytes using the Whole-Blood column kit with the StraightFrom® Whole Blood CD14 MicroBeads in humans (Miltenyi Biotec; Bergisch Gladbach, Germany) according to the manufacturer’s instructions. The isolated DNA was bisulfite-modified with the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA). The samples were sent for library preparation and subsequent WGBS to the Genomic and Proteomics Core Facility of the German Cancer Research Center (DKFZ) in Heidelberg (Germany). The sequencing was performed on the HiSeqX platform (Illumina, San Diego, CA, USA) with one sample per lane. The resulting read data was processed as previously described36,57,58. In brief, reads were aligned against the hg19 reference genome built hg19 using bwa-meth (v0.2.0)59 and subsequently sorted using samtools (v1.3.1)60. After deduplicating the reads with sambamba (v0.6.0)61, quality control applying qualimap (v2.2.2)62 was performed. Basic statistics related to the process success are presented in Supplementary Table 1.
For genome-wide DMR calling, we utilized camel63 and metilene64 following their respective manuals. In the camel analysis, DNA methylation levels were initially called by camel itself, and DMRs were identified based on DNA methylation differences, using a threshold of four CpG sites and a minimum difference of 0.3. As no DMRs were identified at this threshold, we adjusted the parameters to a minimum DNA methylation difference of 0.2 and increased the stringency to a minimum of 10 CpG sites. Additionally, metilene was employed to call DMRs using a significance threshold of q < 0.05. Following DMR identification, we applied PCA using the Python scikit-learn library65 to visualize differences of methylation patterns across sample groups. For the PCA, we included CpG loci that had a minimum coverage of 10 reads in all analysed samples and a minimum mapping quality score of 30. Furthermore, hierarchical clustering with a cityblock (Manhattan) metric was performed using scikit-learn on the 1,000 CpG sites with the highest standard deviation across all samples to further explore differences between samples. CpG SNPs were excluded from this analysis66.
To validate the WGBS-derived DMRs, a DBS approach was performed for eight DMRs (four detected by both DMR calling tools, and four detected by camel only) in the DMR identification cohort. We applied a two-step PCR similar to the one used for the NR1H3 gene locus applying DMR-specific primers (see Supplementary Table 2). PCR products were purified using the QIAquick 96 PCR Purification Kit (QIAGEN, Venlo, Netherlands). The DBS was performed at the Genomics and Transcriptomics Facility at the University Hospital Essen (Germany). We visually analysed whether the studied DMRs replicated technically using the amplikyzer software52.
Data availability
The original contributions presented in the study are publicly available. This data can be found here: https://www.ebi.ac.uk/ena/browser/view/PRJEB38906.
References
Hinney, A., Korner, A. & Fischer-Posovszky, P. The promise of new anti-obesity therapies arising from knowledge of genetic obesity traits. Nat. Rev. Endocrinol. https://doi.org/10.1038/s41574-022-00716-0 (2022).
Udo, T. & Grilo, C. M. Prevalence and correlates of DSM-5-defined eating disorders in a nationally representative sample of US. Adults. Biol. Psychiatry 84, 345–354. https://doi.org/10.1016/j.biopsych.2018.03.014 (2018).
Bulik, C. M. et al. Genetics and neurobiology of eating disorders. Nat. Neurosci. 25, 543–554. https://doi.org/10.1038/s41593-022-01071-z (2022).
American Psychiatric Association. American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders. (2013).
Hebebrand, J. & Bulik, C. M. Critical appraisal of the provisional DSM-5 criteria for anorexia nervosa and an alternative proposal. Int. J. Eat. Disord. 44, 665–678. https://doi.org/10.1002/eat.20875 (2011).
Shimazu, T. et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. https://doi.org/10.1126/science.1227166 (2013).
Dominguez-Salas, P. et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun 5, 3746. https://doi.org/10.1038/ncomms4746 (2014).
Heijmans, B. T. et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 105, 17046–17049. https://doi.org/10.1073/pnas.0806560105 (2008).
Hübel, C., Marzi, S. J., Breen, G. & Bulik, C. M. Epigenetics in eating disorders: A systematic review. Mol. Psychiatry 24, 901–915. https://doi.org/10.1038/s41380-018-0254-7 (2019).
Käver, L. et al. Epigenetic alterations in patients with anorexia nervosa-a systematic review. Mol. Psychiatry https://doi.org/10.1038/s41380-024-02601-w (2024).
Ehrlich, S. et al. Promoter specific DNA methylation and gene expression of POMC in acutely underweight and recovered patients with anorexia nervosa. J. Psychiatr. Res. 44, 827–833. https://doi.org/10.1016/j.jpsychires.2010.01.011 (2010).
Frieling, H. et al. Epigenetic downregulation of atrial natriuretic peptide but not vasopressin mRNA expression in females with eating disorders is related to impulsivity. Neuropsychopharmacology 33, 2605–2609. https://doi.org/10.1038/sj.npp.1301662 (2008).
Frieling, H. et al. Global DNA hypomethylation and DNA hypermethylation of the alpha synuclein promoter in females with anorexia nervosa. Mol. Psychiatry 12, 229–230. https://doi.org/10.1038/sj.mp.4001931 (2007).
Frieling, H. et al. Epigenetic dysregulation of dopaminergic genes in eating disorders. Int. J. Eat. Disord. 43, 577–583. https://doi.org/10.1002/eat.20745 (2010).
Neyazi, A. et al. Association of leptin gene DNA methylation with diagnosis and treatment outcome of anorexia nervosa. Front. Psychiatry 10, 197. https://doi.org/10.3389/fpsyt.2019.00197 (2019).
Boehm, I. et al. Peripheral serotonin transporter DNA methylation is linked to increased salience network connectivity in females with anorexia nervosa. J. Psychiatry Neurosci. 45, 8. https://doi.org/10.1503/jpn.190016 (2019).
Sild, M. & Booij, L. Histone deacetylase 4 (HDAC4): A new player in anorexia nervosa?. Mol. Psychiatry 24, 1425–1434. https://doi.org/10.1038/s41380-019-0366-8 (2019).
Subramanian, S., Braun, P. R., Han, S. & Potash, J. B. Investigation of differential HDAC4 methylation patterns in eating disorders. Psychiatr. Genet. 28, 12–15. https://doi.org/10.1097/YPG.0000000000000189 (2018).
Thaler, L. et al. Methylation of the OXTR gene in women with anorexia nervosa: Relationship to social behavior. Eur. Eat Disord. Rev. 28, 79–86. https://doi.org/10.1002/erv.2703 (2020).
Booij, L. et al. DNA methylation in individuals with anorexia nervosa and in matched normal-eater controls: A genome-wide study. Int. J. Eat. Disord. 48, 874–882. https://doi.org/10.1002/eat.22374 (2015).
Kesselmeier, M. et al. High-throughput DNA methylation analysis in anorexia nervosa confirms TNXB hypermethylation. World J. Biol. Psychiatry 19, 187–199. https://doi.org/10.1080/15622975.2016.1190033 (2018).
Steiger, H. et al. A longitudinal, epigenome-wide study of DNA methylation in anorexia nervosa: Results in actively ill, partially weight-restored, long-term remitted and non-eating-disordered women. J. Psychiatry Neurosci. 44, 205–213. https://doi.org/10.1503/jpn.170242 (2019).
Steiger, H. et al. DNA methylation in people with anorexia nervosa: Epigenome-wide patterns in actively ill, long-term remitted, and healthy-eater women. World J. Biol. Psychiatry 24, 254–259. https://doi.org/10.1080/15622975.2022.2089731 (2023).
Steiger, H. et al. Elevated plasma B(12) and betaine levels in women with anorexia nervosa: Possible role in illness pathophysiology and epigenetic regulation. J. Psychiatry Neurosci. 50, E85–E91. https://doi.org/10.1503/jpn.240155 (2025).
Farre, P. et al. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenet. Chromatin. 8, 19. https://doi.org/10.1186/s13072-015-0011-y (2015).
Loyfer, N. et al. A DNA methylation atlas of normal human cell types. Nature https://doi.org/10.1038/s41586-022-05580-6 (2023).
COvid-19 Multi-omics Blood ATlas Consortium. A blood atlas of COVID-19 defines hallmarks of disease severity and specificity. Cell, 185, 916–e958 (2022). https://doi.org/10.1016/j.cell.2022.01.012
Nowak, J., Borkowska, B. & Pawlowski, B. Leukocyte changes across menstruation, ovulation, and mid-luteal phase and association with sex hormone variation. Am. J. Hum. Biol. 28, 721–728. https://doi.org/10.1002/ajhb.22856 (2016).
Cole, S. W. Elevating the perspective on human stress genomics. Psychoneuroendocrinology 35, 955–962. https://doi.org/10.1016/j.psyneuen.2010.06.008 (2010).
Jones, M. J., Moore, S. R. & Kobor, M. S. Principles and challenges of applying epigenetic epidemiology to psychology. Annu. Rev. Psychol. 69, 459–485. https://doi.org/10.1146/annurev-psych-122414-033653 (2018).
Iranzo-Tatay, C. et al. Genome-wide DNA methylation profiling in anorexia nervosa discordant identical twins. Transl. Psychiatry 12, 15. https://doi.org/10.1038/s41398-021-01776-y (2022).
Horsthemke, B. A critical appraisal of clinical epigenetics. Clin. Epigenetics 14, 95. https://doi.org/10.1186/s13148-022-01315-6 (2022).
Reinius, L. E. et al. Differential DNA methylation in purified human blood cells: Implications for cell lineage and studies on disease susceptibility. PLoS ONE 7, e41361. https://doi.org/10.1371/journal.pone.0041361 (2012).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf. 13, 86. https://doi.org/10.1186/1471-2105-13-86 (2012).
Ziller, M. J., Hansen, K. D., Meissner, A. & Aryee, M. J. Coverage recommendations for methylation analysis by whole-genome bisulfite sequencing. Nat. Methods 12, 230–232. https://doi.org/10.1038/nmeth.3152 (2015).
Wallner, S. et al. Epigenetic dynamics of monocyte-to-macrophage differentiation. Epigenet. Chromatin 9, 33. https://doi.org/10.1186/s13072-016-0079-z (2016).
Freff, J. et al. Alterations in B cell subsets correlate with body composition parameters in female adolescents with anorexia nervosa. Sci. Rep. 11, 1125. https://doi.org/10.1038/s41598-020-80693-4 (2021).
Föcker, M. et al. Comparison of metabolic profiles of acutely ill and short-term weight recovered patients with anorexia nervosa reveals alterations of 33 out of 163 metabolites. J. Psychiatr. Res. 46, 1600–1609. https://doi.org/10.1016/j.jpsychires.2012.08.015 (2012).
Föcker, M. et al. Evaluation of metabolic profiles of patients with anorexia nervosa at inpatient admission, short- and long-term weight regain-descriptive and pattern analysis. Metabolites 11, 7. https://doi.org/10.3390/metabo11010007 (2020).
Christiansen, L. et al. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell 15, 149–154. https://doi.org/10.1111/acel.12421 (2016).
Tan, Q. et al. Epigenetic drift in the aging genome: A ten-year follow-up in an elderly twin cohort. Int. J. Epidemiol. 45, 1146–1158. https://doi.org/10.1093/ije/dyw132 (2016).
Duan, R., Fu, Q., Sun, Y. & Li, Q. Epigenetic clock: A promising biomarker and practical tool in aging. Ageing Res. Rev. 81, 101743. https://doi.org/10.1016/j.arr.2022.101743 (2022).
Ciccarone, F., Tagliatesta, S., Caiafa, P. & Zampieri, M. DNA methylation dynamics in aging: How far are we from understanding the mechanisms?. Mech. Ageing Dev. 174, 3–17. https://doi.org/10.1016/j.mad.2017.12.002 (2018).
Refn, M. R. et al. Longitudinal changes and variation in human DNA methylation analysed with the Illumina MethylationEPIC BeadChip assay and their implications on forensic age prediction. Sci. Rep. 13, 21658. https://doi.org/10.1038/s41598-023-49064-7 (2023).
Sellami, M., Bragazzi, N., Prince, M. S., Denham, J. & Elrayess, M. Regular, intense exercise training as a healthy aging lifestyle strategy: Preventing DNA damage, telomere shortening and adverse DNA methylation changes over a lifetime. Front. Genet. 12, 652497. https://doi.org/10.3389/fgene.2021.652497 (2021).
Nicoletti, C. F. et al. DNA methylation pattern changes following a short-term hypocaloric diet in women with obesity. Eur. J. Clin. Nutr. 74, 1345–1353. https://doi.org/10.1038/s41430-020-0660-1 (2020).
American Psychological Association. Diagnostic and statistical manual of mental disorders 4th edn. (American Psychiatric Publishing Inc, 1994).
Kaufman, J. et al. Schedule for affective disorders and schizophrenia for school-age children-present and lifetime version (K-SADS-PL): Initial reliability and validity data. J. Am. Acad. Child. Adolesc. Psychiatry 36, 980–988. https://doi.org/10.1097/00004583-199707000-00021 (1997).
Herpertz-Dahlmann, B. Treatment of eating disorders in child and adolescent psychiatry. Curr. Opin .Psychiatry 30, 438–445. https://doi.org/10.1097/YCO.0000000000000357 (2017).
World Medical Association. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 310, 2191–2194. https://doi.org/10.1001/jama.2013.281053 (2013).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16, 1215. https://doi.org/10.1093/nar/16.3.1215 (1988).
Rahmann, S. et al. Amplikyzer: Automated methylation analysis of amplicons from bisulfite flowgram sequencing. Report No. 2167–9843, (PeerJ PrePrints, 2013).
Austermann, J., Roth, J. & Barczyk-Kahlert, K. The good and the bad: Monocytes’ and Macrophages’ Diverse Functions in Inflammation. Cells https://doi.org/10.3390/cells11121979 (2022).
Nilsson, I. A. K. et al. Aberrant inflammatory profile in acute but not recovered anorexia nervosa. Brain Behav. Immun. 88, 718–724. https://doi.org/10.1016/j.bbi.2020.05.024 (2020).
Zhu, Y. et al. Genome-wide profiling of DNA methylome and transcriptome in peripheral blood monocytes for major depression: A Monozygotic Discordant Twin Study. Transl Psychiatry 9, 215. https://doi.org/10.1038/s41398-019-0550-2 (2019).
Biemann, R. et al. Gene expression profile of CD14(+) blood monocytes following lifestyle-induced weight loss in individuals with metabolic syndrome. Sci. Rep. 10, 17855. https://doi.org/10.1038/s41598-020-74973-2 (2020).
Rademacher, K. et al. Evolutionary origin and methylation status of human intronic CpG islands that are not present in mouse. Genome Biol. Evol. 6, 1579–1588. https://doi.org/10.1093/gbe/evu125 (2014).
Hummel, E. et al. No evidence for intervention-associated DNA methylation changes in monocytes of patients with posttraumatic stress disorder. Sci. Rep. 12, 17347. https://doi.org/10.1038/s41598-022-22177-1 (2022).
Pedersen, B. S., Eyring, K., De, S., Yang, I. V. & Schwartz, D. A. Fast and accurate alignment of long bisulfite-seq reads. arXiv preprint arXiv:1401.1129 (2014).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience https://doi.org/10.1093/gigascience/giab008 (2021).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. https://doi.org/10.1093/bioinformatics/btv098 (2015).
Okonechnikov, K., Conesa, A. & Garcia-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. https://doi.org/10.1093/bioinformatics/btv566 (2016).
Schröder, C. Bioinformatics from genetic variants to methylation PhD thesis, (2018).
Juhling, F. et al. metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 26, 256–262. https://doi.org/10.1101/gr.196394.115 (2016).
Varoquaux, G. et al. Scikit-learn: Machine learning without learning the machinery. GetMobile Mobile Comput. Commun. 19, 29–33. https://doi.org/10.1145/2786984.2786995 (2015).
Schröder, C. et al. Regions of common inter-individual DNA methylation differences in human monocytes: Genetic basis and potential function. Epigenet. Chromatin 10, 37. https://doi.org/10.1186/s13072-017-0144-2 (2017).
Acknowledgements
We thank all participants for their participation in our study. Further, we are indebted to Sieglinde Düerkop for her excellent technical support. This study was funded by the Deutsche Forschungsgemeinschaft (DFG, HI 865/2-1, DFG Research Unit FOR2488), the BMBF (01GS0820; PALGER 2017-33: 01DH19010) and the Stiftung Universitätsmedizin Essen. We acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MK, JH, BH, AH and LSR conceptualized the study. MF, IK, BHD, JS, MdZ, WH, SE, SZ, KG, KE, RB, JH and AH recruited participants and provided samples. CS1, CS2, TP, MF, IK and LSR curated the data. MK, CS1, CS2, TP, JB, EL, MZ, JG, BB, BH, AH and LSR performed the analyses and interpreted the results. MK, AH and LSR wrote the manuscript. All authors reviewed the manuscript and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rajcsanyi, L.S., Kesselmeier, M., Schröder, C. et al. No indications of weight gain associated DNA methylation changes in patients with anorexia nervosa. Sci Rep 15, 28870 (2025). https://doi.org/10.1038/s41598-025-12592-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12592-5
