
Abstract
Macrophage senescence is closely associated with the progression of periodontitis. In vitro investigations have demonstrated that the colony-stimulating factor 1 receptor (CSF1R) inhibitor, PLX3397, reduces macrophage senescence; however, the underlying mechanisms remain unclear. This study explored whether CSF1R inhibitors mitigate periodontitis by reducing macrophage senescence and examined the role of the PI3K/AKT/FOXO1 signaling pathway. Using C57BL/6 mice and RAW264.7 cells, periodontal tissues were analyzed with Micro-CT, hematoxylin and eosin staining, immunofluorescence, and immunohistochemistry. Senescence-associated-β-galactosidase staining, RT-qPCR, and western blotting assessed cellular senescence, while reactive oxygen species (ROS) levels were measured by flow cytometry. Our findings revealed that PLX3397 significantly reduced periodontal tissue destruction and inflammation, and decreased the number of senescent macrophages in a murine periodontitis model. In Porphyromonas gingivalis-lipopolysaccharide-stimulated RAW264.7 macrophages, PLX3397 reduced macrophage senescence via PI3K/AKT/FOXO1 signaling and downregulated ROS expression. These findings demonstrate that CSF1R inhibitors effectively mitigate periodontitis by targeting macrophage senescence through the PI3K/AKT/FOXO1 pathway, providing potential therapeutic insights for periodontitis.
Similar content being viewed by others


Introduction
Periodontitis is a chronic inflammatory condition affecting the periodontal supporting tissues, which affects nearly two-thirds of the global population1. This disease is strongly associated with systemic conditions, including diabetes mellitus, cardiovascular disease, and Alzheimer’s disease2,3,4. The progression of periodontitis is attributed to an imbalance between microbial plaque accumulation and the host’s inflammatory response5. Although bacteria are the primary etiological agents of periodontitis, bacterial infection alone cannot drive disease progression6. Instead, the host’s inflammatory response to bacterial infection alters the gingival microenvironment, thereby destroying periodontal tissues7. Recent evidence has identified senescent cells within the periodontal microenvironment as a significant source of chronic inflammation, contributing to the ongoing destruction of periodontal tissues8,9. Contemporary literature suggests that periodontitis involves a “dual source” of inflammation, driven by both the initial bacterial infection and senescence-associated inflammation resulting from the accumulation of senescent cells8. Identifying these senescent cells offers novel insights into the mechanisms underlying periodontal tissue degradation10,11,12.
Macrophages, a critical component of the host immune response, are differentiated from monocytes into mature macrophages upon exposure to external stimuli. Under physiological conditions, macrophages are pivotal in maintaining homeostasis in vivo. Conversely, in pathological states, factors such as DNA damage, telomere shortening, oncogene activation, and various stressors activate senescence-associated signaling pathways, including the p53 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways, thereby inducing macrophage senescence13. A critical characteristic of senescent cells is the development of a senescence-associated secretory phenotype (SASP), which comprises a maladaptive assemblage of proinflammatory cytokines and matrix-degrading proteases that collectively alter the surrounding microenvironment. Macrophages are central contributors to the secretion of SASP14. Senescent macrophages secrete substantial quantities of SASP factors within the gingival microenvironment. This secretion serves dual purposes: enhancing and propagating cellular senescence, which increases the burden of senescent cells and SASP production while amplifying the release of inflammatory mediators and facilitating the activation and infiltration of additional inflammatory cells such as neutrophils. Consequently, these processes contribute to ongoing tissue destruction15. As senescence progresses, the late-stage SASP response appears to worsen periodontal lesions. This low-grade inflammation triggers further SASP activation in macrophages. Increased senescence of gingival macrophages induces bacterial alterations that enhance their inflammatory properties16. Therefore, targeting macrophage senescence by inhibiting or selectively removing senescent macrophages presents a promising therapeutic strategy for mitigating the exacerbated destruction of periodontal tissues observed during the progression of periodontitis. However, the underlying mechanisms driving macrophage senescence in periodontitis remain unclear.
Cellular senescence is the irreversible cessation of cell cycle progression in response to various damaging and stressful stimuli17, such as DNA damage, oxidative stress, and exogenous inflammatory agents18. The onset of cellular senescence is believed to be caused by oxidative stress19, with the accumulation of reactive oxygen species (ROS) identified as a direct factor facilitating oxidative stress within an organism20. Periodontitis is an inflammatory disease characterized by oxidative stress, leading to excessive ROS generation21. The PI3K/AKT signaling pathway orchestrates the critical transition between immune activation and suppression during inflammatory responses22 and can be activated by ROS23. FOXO1, a pivotal downstream target of the PI3K/AKT pathway, also plays a significant role in ROS regulation24.
The colony-stimulating factor 1 receptor (CSF1R) is essential for macrophage differentiation, survival, proliferation, adhesion, and migration. Overactivation of CSF1R can result in the overexpression of proinflammatory cytokines, including TNF-α and IL-625. PLX3397, a selective adenosine triphosphate (ATP)-competitive inhibitor of CSF1R, exhibits a high affinity for CSF1R, reduces macrophage survival and migration, and inhibits CSF1R signaling26,27,28. PLX3397 has been extensively used in clinical settings for treating tenosynovial giant cell tumors and has demonstrated favorable outcomes29. Additionally, one study indicated that PLX3397 administration during the induction phase of periodontitis mitigated disease severity in both aged and young murine models30. Our previous study demonstrated that PLX3397 effectively inhibited lipopolysaccharide (LPS)-induced senescence in bone marrow-derived macrophages31. Therefore, elucidating the specific mechanisms by which CSF1R inhibitors regulate macrophage senescence to modulate inflammation in periodontitis would be advantageous for optimizing host inflammatory responses and mitigating periodontal tissue destruction.
Currently, the treatment of periodontitis mainly relies on local mechanical removal of plaque and calculus. However, due to factors such as periodontal pockets or anatomical structures, plaque removal may be incomplete in some patients, which further worsens periodontal conditions32. Therefore, it is necessary to combine traditional methods with immunotherapy and other approaches in the treatment of periodontitis. According to our hypothesis, CSF1R inhibitors decrease periodontitis by reducing PI3K/AKT/FOXO1 mediated macrophage senescence. In this study, we demonstrated through animal experiments that CSF1R inhibitors reduce macrophage senescence and alleviate periodontal inflammation. Further cellular experiments explored the potential mechanisms by which CSF1R inhibitors regulate macrophage senescence. Our findings provide a theoretical basis for future clinical approaches to alleviate periodontal inflammation by modulating the host immune response.
Materials and methods
Mouse model
An Animal Ethics Committee at Guizhou Medical University approved all animal experiments (Accession Number: 2200991) and followed the university’s guidelines for the care and use of experimental animals, as well as the ARRIVE 2.0 guidelines. Thirty male C57BL/6 wild-type mice, aged 7 weeks, were randomly divided into three groups: normal (N, n = 10), periodontitis (PD, n = 10), and treatment (PLX, n = 10). These mice were housed in Guizhou Medical University’s animal facility under standard conditions, with unrestricted access to water and food.
Following a 1-week acclimation period, the mice were anesthetized using tribromoethanol (0.2 mL/10 g; Nanjing Aibei Biotech, Nanjing, China). To create a periodontitis model, a 5 − 0 silk ligature was fastened around the mandibular first molar for 2 weeks. Throughout the disease induction period, mice in the treatment group received daily intragastric administration of pexidartinib (PLX3397; MedChemExpress, Shanghai, China) at 50 mg/kg. The doses administered to the animals were referenced from previous studies30.
Micro-CT analysis
After removing the surrounding soft tissues, the mandibular jaws from each group were scanned using micro-computed tomography (micro-CT/µCT) with a 10 μm resolution. Alveolar bone loss was assessed through morphometric examination of the mandibular micro-CT images. On the lingual side of the mandibular first molars, the cemento-enamel junction, alveolar bone crest, and mesial and distal line angles were measured to determine the degree of alveolar bone loss. To analyze bone mineral density (BMD), a rectangular area that included the loss of alveolar bone was designated as the volume of interest.
Hematoxylin and Eosin staining
Mouse mandibles were decalcified in 10% ethylenediaminetetraacetic acid solution, which was refreshed every 2 days over a 3-week period. The specimens were decalcified, immersed in paraffin, and then divided into slices with a thickness of 5 mm. Following two hours of deparaffinization at 65 °C, the sections were dehydrated with Xylene and graded ethanol series. Subsequently, the sections were stained using a hematoxylin and eosin (H&E) kit (Servicebio, Wuhan, China). Stained images were captured using a light microscope equipped with a digital camera (NIKON DS-U3; Nikon Instruments Inc., Tokyo, Japan). The width of the periodontal ligament was measured using ImageJ software, which measures the distance from the furcation of the mandibular first molar’s roots to the interradicular septum’s apex.
Immunohistochemical staining
Paraffin-embedded sections of mouse tissues were deparaffinized, hydrated, and treated with a 3% hydrogen peroxide solution in phosphate-buffered saline (PBS) for 10 min to block endogenous peroxidase activity. After antigen retrieval, samples were incubated with 10% goat serum in PBS for 30 min. The sections were stained using antibodies targeting CSF1R (1:100; YT0881; Immunoway, Plano, TX, USA), IL-6 (1:100; YT5348; Immunoway, USA), IL-1β (1:100; YT5201; Immunoway, USA), and TNF-α (1:100; YT4689; Immunoway, USA). Following three PBS washes, the sections were incubated with goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (1:200; GB23303; Servicebio, China) for 30 min at ambient temperature. Immunohistochemical staining was conducted using an immunohistochemical kit (Servicebio, China) with a DAB chromogenic agent. Digital cameras (NIKON DS-U3, Japan) were used to capture stained images.
Immunofluorescent staining
Deparaffinized and hydrated mouse tissues were used for heat-induced antigen retrieval using citrate buffer (pH 6.0). After three 5-minute washes with PBS, the sections were incubated for 15 min in a solution containing 0.3% Triton X−100 and 2% goat serum in PBS. Following three additional washes with PBS, the sections were incubated for 30 min with 10% goat serum in PBS. Primary antibodies targeting F4/80 (1:50; RT1212; Huabio, Hangzhou, China), p16 (1:50; ET1602-9; Huabio, China), and p21 (1:50; HA500005; Huabio, China) were applied overnight at 4 °C. Incubation with a goat anti-rat fluorescein isothiocyanate (FITC)-conjugated secondary antibody (1:200; Servicebio, China) specifically bound to F4/80 was performed after three 20-min PBS washes. Additionally, a goat anti-rabbit CY3-conjugated secondary antibody (1:300; GB21303; Servicebio, China), targeting p16 and p21, was used during incubation. After a 1-h incubation period, the sections were rinsed and stained with DAPI. Digital cameras (NIKON DS-U3, Japan) were used to capture stained images.
Cells and treatments
RAW264.7 cells (FuHeng Biology, Shanghai, China) were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum at 37 °C in a 5% CO2 incubator. To establish the in vitro periodontitis model, the cells were stimulated with 1 µg/mL of Porphyromonas gingivalis (P.g)-derived LPS (InvivoGen, Toulouse, France) 24 h. The timing and concentration of LPS used to induce macrophage senescence were determined based on previous studies33,34. Before the LPS priming stage, pexidartinib (PLX3397; MedChemExpress, China) was introduced to the cultures at concentrations of 100 nM, 500 nM, and 1000 nM. In order to study the effects of PI3K inhibitors, the cells were pre-stimulated with LY294002 (MedChemExpress, China) at concentrations of 25 µM prior 4 h to LPS stimulation. We collected the total mRNA and protein content after 24 h of LPS stimulation.
Senescence-associated-β-galactosidase staining
Senescence-associated -β-galactosidase (SA–β–gal) staining was conducted using the SA–β–gal staining kit (Beyotime Biotech, Shanghai, China), following the manufacturer’s instructions. Cells were rinsed thrice with PBS and then fixed with 4% paraformaldehyde (PFA) at room temperature for 15 min. After fixation, the cells were incubated overnight at 37 °C in the dark with a solution containing 0.05 mg/mL of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside. Blue color formation was evaluated under a microscope at a 20× magnification after incubation using PBS rinsed with PBS. Images were acquired using a light microscope equipped with a digital camera (NIKON DS-U3, Japan).
RT-qPCR analysis
Total RNA was extracted from RAW264.7 cells using an RNA-Quick Purification Kit (YiShan Biotech, Shanghai, China). Subsequently, RNA was reverse-transcribed into cDNA using the Fast All-in-One RT Kit (gDNA Remover; YiShan Biotech, China) according to the manufacturer’s instructions. The SYBR Green PCR Master Mix (Vazyme, Nanjing, China) was used for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis. The housekeeping gene, GAPDH, was used as a control, and relative expression levels of the target mRNAs were quantified using the comparative 2−ΔΔCT method. The primers used were as follows:
IL-6: 5’-CTTCTTGGGACTGATGCTGGT-3’ (Forward);
and 5’-AGGTCTGTTGGGAGTGGTATCC-3’ (Reverse).
IL-1β: 5’-AGCTTCAGGCAGGCAGTATC-3’(Forward);
and 5’-AAGGTCCACGGGAAAGACAC-3’(Reverse).
TNF-α: 5’-ATGAGCACAGAAAGCATGATC-3’(Forward);
and 5’-TACAGGCTTGTCACTCGAATT-3’(Reverse).
GAPDH: 5’-GGTTGTCTCCTGCGACTTCA-3’(Forward);
and 5’-TGGTCCAGGGTTTCTTACTCC-3’(Reverse).
Western blotting
A total protein extraction kit was used to extract proteins from RAW264.7 cells in accordance with the manufacturer’s instructions. Protein concentrations were determined using a bicinchoninic acid (BCA) assay. Protein samples were fractionated using sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. After blocking with 5% skimmed milk in Tris-buffered saline with Tween (TBST) for 1 h, the membranes were incubated overnight at 4 °C with primary antibodies, including CSF1R (1:1,500; YT0881; Immunoway, USA), PI3K (1:1,500; YM3503; Immunoway, USA), phosphorylated PI3K (p-PI3K) (1:1,000; AF3241; Affinity, USA), AKT (1:1,500; YT0185; Immunoway, USA), phosphorylated-AKT (p-AKT) (1:1,000; YP0006; Immunoway, USA), FOXO1 (1:1,500; YT1758; Immunoway, USA), phosphorylated-FOXO1 (p-FOXO1) (1:1,000; YP0113; Immunoway, USA), p16 (1:1,500; ET1602-9; Huabio, China), p21 (1:1,500; HA500005; Huabio, China), and β-actin (1:10,000; YM3028; Immunoway, USA). After washing with TBST, incubation at room temperature for 1 h with secondary antibodies against rabbit (1:5,000; HA1001; Huabio, China) or mouse (1:5,000; HA1006; Huabio, China) was performed. The gel imaging system GeneGnome (SYNGENE, Cambridge, USA) was used to measure the signal intensities of protein bands identified using a boosted chemiluminescence kit. Blot quantification was performed using ImageJ software.
Flow cytometry
Cellular ROS levels were evaluated using a Reactive Oxygen Species assay kit (Solarbio, Beijing, China). A 10 mM solution of 2′, 7′-Dichlorodihydrofluorescein diacetate (DCFH‐DA) was prepared and diluted in a serum-free medium to produce a working solution of DCFH‐DA with a 10 µM concentration. After washing with PBS, the DCFH-DA working solution was used to incubate the cells for 20 min at 37 °C. The cells were collected for flow cytometry analysis after three washes with serum-free medium. Results were analyzed using FlowJo (version 10.8.1; FlowJo LLC, Ashland, Oregon, USA). Relative ROS level was calculated according to the following formula:
whereby MFItest group represents the mean fluorescence intensity (MFI) of ROS measured in drug treated groups (P.g-LPS group, PLX3397 group or LY294002 group), MFIcontrol group represents the MFI of ROS measured in the control group without drugs, and MFIblank group represents the MFI of ROS measured in the blank group without cells and drugs.
Statistical analysis
All experiments were conducted at least three times. Data are presented as mean ± standard error. Statistical analyses were performed using the GraphPad Prism software (version 8.0.0). Students’ t-test or analysis of variance (ANOVA) were used to determine significance. P < 0.05 was set as the level of statistical significance.
Results
PLX3397 alleviates periodontal tissue destruction in mice with periodontitis
To induce periodontitis in mice, a 5 − 0 silk ligature was placed around the first mandibular molar, thereby facilitating bacterial overgrowth and creating a periodontitis model. Following the establishment of the periodontitis model, in vivo experiments were conducted (Fig. 1A). To evaluate the potential efficacy of CSF1R inhibitors in mitigating tissue destruction, PLX3397 was administered immediately after model induction. Micro-CT analysis revealed significantly greater alveolar bone resorption in the periodontitis group compared to the normal group, with a marked reduction in bone density (Fig. 1B). The H&E staining of the periodontal ligament spaces showed a pronounced expansion in the periodontitis group, as compared to the normal group (Fig. 1C). Our experimental model is validated by these observations which are consistent with the characteristics of periodontitis. Mice treated with PLX3397 exhibited significant improvements in periodontal conditions (Figs. 1B and C), indicating that CSF1R inhibitors can effectively mitigate periodontal tissue destruction in murine models of induced periodontitis.

PLX3397 treatment in mice with induced periodontitis resulted in reduced destruction of alveolar bone and periodontal ligament. (A) The experimental periodontitis mouse model was strictly followed, along with all protocols. (B) Reconstructions of the mandibular alveolar bones in three dimensions and measurements of bone mineral density (BMD) in the area of interest (alveolar bone of the mandibular first molars); Scale Bar = 1 mm. (C) Periodontal tissue stained with hematoxylin and eosin. The black areas indicate the periodontal ligament, and the yellow box indicates the periodontal tissue; Scale Bar = 500 μm. The width of the periodontal ligament was measured from the point where the tooth root splits into two branches to the highest point of the alveolar bone; Scale Bar = 100 μm. N = normal mice, PD = mice with periodontitis, PLX = mice with periodontitis treated with PLX3397 at 50 mg/kg. The results are presented as a mean ± standard error; n = 3; comparisons between PD vs. N and PLX vs. PD by one-way ANOVA; **p < 0.01 and ***p < 0.001.
CSF1R Inhibition alleviates inflammation in mice with periodontitis
Periodontal infection triggers the innate immune response, resulting in macrophage infiltration and accumulation at the site of inflammation to identify and eliminate pathogens. In chronic diseases, continuous macrophage infiltration causes a heightened inflammatory state that ultimately results in tissue destruction. PLX3397, a small-molecule CSF1R inhibitor, prevents the differentiation of monocytes into mature macrophages. Immunohistochemical analysis showed that CSF1R expression was upregulated in the periodontitis group, but it was significantly downregulated in the PLX3397-treated group, compared to the normal group (Fig. 2A). H&E staining demonstrated that fibroblasts in the periodontium of the normal group exhibited an orderly arrangement and normal cytoplasmic morphology. In contrast, fibroblasts in the periodontitis group displayed a disorganized arrangement and altered cytoplasmic morphology, indicating heightened inflammation (Fig. 2B). Additionally, the gingival tissues had significantly elevated proinflammatory cytokine levels, specifically IL-6, IL-1β, and TNF-α (Fig. 2C). PLX3397 treatment led to a notable decrease in cytokine (IL-6, IL-1β, and TNF-α) levels and a significant reduction in inflammatory cell numbers (Fig. 2B and C). These findings suggest that attenuating periodontitis-related inflammation in mice may be facilitated by downregulating CSF1R expression.

Reducing CSF1R expression ameliorates inflammatory response in mice with periodontitis. (A) Mandibular gingival tissue immunohistochemical staining for CSF1R and the percentage of CSF1R-positive cells; Scale Bar = 50 μm. (B) Periodontal tissue stained with hematoxylin and eosin. Yellow arrows indicate inflammatory cells; Scale Bar = 50 μm. (C) Mandibular gingival tissue immunohistochemistry staining for IL-6, IL-1β, and TNF-α and the percentage of positive cells; Scale Bar = 50 μm. N = normal mice, PD = mice with periodontitis, PLX = mice with periodontitis treated with PLX3397 at 50 mg/kg. The results are presented as a mean ± standard error; n = 3; comparisons between PD vs. N and PLX vs. PD by one-way ANOVA; ***p < 0.001.
In vivo and in vitro periodontitis models, Inhibition of CSF1R expression reduces macrophage senescence
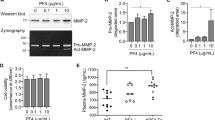
Senescent cells in gingival tissues were identified using the cell cycle inhibitory proteins p16 as markers. Immunofluorescence analysis revealed a significant increase in senescent cells in the periodontitis group (Figs. 3A). Macrophages and senescent macrophages in the gingiva were assessed using the F4/80 marker. The immunofluorescence results indicated a marked increase in both total and senescent macrophages in the periodontitis group, whereas the PLX3397-intervention group exhibited a significant reduction in these cell populations (Fig. 3A). To elucidate the potential of CSF1R expression reduction in alleviating macrophage senescence associated with periodontitis, we developed an in vitro model of macrophage inflammatory senescence. This was achieved by stimulating RAW264.7 cells with LPS-derived from P.g for 24 h. Subsequent SA-β-Gal staining demonstrated that treatment with PLX3397 mitigated macrophage senescence, with 500 nM identified as the optimal concentration (Figs. 3B and C). Furthermore, WB analysis demonstrated that inhibition of CSF1R was most pronounced at 500 nM PLX3397, which effectively downregulated the expression of p16 and p21 proteins (Fig. 3D). An RT-qPCR analysis showed that treatment with 500 nM PLX3397 decreased IL-6, IL-1β, and TNF-α expression (Fig. 3E), all of which are SASP components. These results suggest that inhibiting CSF1R expression can mitigate macrophage senescence, with 500 nM identified as the optimal inhibitory concentration.

Reducing CSF1R expression decreases macrophage senescence in both in vivo and in vitro periodontitis experiments. (A) Mandibular gingival tissue stained with immunofluorescence for p16 and F4/80. The region above the white dotted line represents the gingival epithelial layer, while the region below corresponds to the subepithelial layer; white arrows indicate cells that are double-positive for p16 and F4/80; Scale Bar = 50 μm. (B) SA-β-gal activity in RAW264.7 cells across groups; Scale Bar = 100 μm. (C) The proportion of SA-β-gal positive cells across groups. (D) The protein expression of CSF1R, p16, p21, and β-actin in RAW264.7 cells, and the values of CSF1R, p16, and p21 levels relative to β-actin across groups. (E) The mRNA expression of IL-6, IL-1β, and TNF-α relative to GAPDH across groups. N = normal mice, PD = mice with periodontitis, PLX = mice with periodontitis treated with PLX3397 at 50 mg/kg. C = control cells, LPS = cells treated with P.g-LPS, L = cells treated with 100 nM PLX3397, M = cells treated with 500 nM PLX3397, H = cells treated with 1,000 nM PLX3397. The results are presented as a mean ± standard error; n = 3; comparisons between PD vs. N, PLX vs. PD, C vs. LPS, L vs. LPS, M vs. LPS and H vs. LPS by one-way ANOVA; *p < 0.05, **p < 0.01, and ***p < 0.001.
In vitro experiments indicate that CSF1R inhibitors reduce macrophage senescence via the PI3K/AKT/FOXO1 signaling pathway
To confirm that CSF1R inhibitors mitigate macrophage senescence through the PI3K/AKT/FOXO1 pathway, supplementary experiments were conducted using the PI3K inhibitor, LY294002, combined with the efficacious concentration of PLX3397 (Fig. 4A). Results from SA-β-Gal staining, WB, and RT-qPCR demonstrated that the PI3K inhibitor reduced macrophage senescence (Figs. 4B–D). Furthermore, WB analysis revealed that total protein levels of PI3K, AKT, and FOXO1 in senescent macrophages remained stable compared to the control group. However, senescent macrophages demonstrated a significant increase in phosphorylated protein levels (Fig. 4E), suggesting the activation of the PI3K/AKT/FOXO1 pathway. Treatment with a PI3K inhibitor notably decreased the expression of phosphorylated PI3K, AKT, and FOXO1 proteins without altering total protein levels (Fig. 4E). WB analysis further indicated that the optimal concentration of the CSF1R inhibitor effectively modulated the PI3K/AKT/FOXO1 signaling pathway (Fig. 4E). These findings suggest that CSF1R inhibitors attenuate macrophage senescence by modulating the PI3K/AKT/FOXO1 pathway.

PLX3397 reduces macrophage senescence via PI3K/AKT/FOXO1 signaling. (A) The periodontitis model in vitro, and all protocols followed the procedure exactly. (B) SA-β-gal activity in RAW264.7 cells, and the proportion of SA-β-gal positive cells across groups; Scale Bar = 100 μm. (C) The protein expression of p16 and p21 in RAW264.7 cells and the values of p16 and p21 levels normalized to β-actin across groups. (D) The mRNA expression of IL-6, IL-1β, and TNF-α relative to GAPDH across groups. (E) The protein expression of p-PI3K, PI3K, p-AKT, AKT, p-FOXO1, FOXO1, p16, p21, and β-actin in RAW264.7 cells and the values of PI3K, AKT and FOXO1 levels relative to β-actin and p-PI3K/PI3K, p-AKT/AKT, and p-FOXO1/FOXO1 across groups. C = control cells, LPS = cells treated with P.g-LPS, M = cells treated with 500 nM PLX3397, and LY = cells treated with 25 µM LY294002. The results are presented as a mean ± standard error; n = 3; comparisons between C vs. LPS, M vs. LPS, and LY vs. LPS by one-way ANOVA; *p < 0.05, **p < 0.01, and ***p < 0.001.
CSF1R and PI3K inhibitors can reduce ROS expression
There is a causal relationship between ROS accumulation and macrophage senescence. Our study confirmed these findings using flow cytometry analysis, which showed increased ROS levels in senescent macrophages. Treatment with the CSF1R inhibitor, PLX3397, and the PI3K inhibitor, LY294002, significantly reduced ROS levels (Fig. 5). These results suggest that CSF1R inhibitors may attenuate ROS production by targeting the PI3K/AKT/FOXO1 signaling pathway, thereby alleviating macrophage senescence.

A flow cytometry analysis was used to detect MFI of ROS with FITC/ROS-A dyeing. C = control cells, LPS = cells treated with P.g-LPS, M = cells treated with 500 nM PLX3397, and LY = cells treated with 25 µM LY294002. The results are presented as a mean ± standard error; n = 3; comparisons between C vs. LPS, M vs. LPS, and LY vs. LPS by one-way ANOVA; *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
Periodontitis creates an environment conducive to cellular senescence, characterized by an accumulation of senescent cells within the periodontal tissues. This phenomenon is driven by four primary factors: persistent gram-negative bacterial infections, chronic inflammation, continuous regeneration of damaged tissue, and localized immunosuppression induced by bacteria35. Through immunofluorescence staining for the senescence markers, p16, we observed a significant increase in senescent cells in mice with periodontitis. Similarly, the macrophage surface marker, F4/80, revealed a substantial rise in senescent macrophages within the inflamed periodontal environment. Although macrophages are terminally differentiated and do not undergo replicative senescence, they are susceptible to stress-induced senescence. This senescence-induced impairment of immune cell function, particularly in macrophages, can precipitate immunodeficiency, thereby reducing immune surveillance and phagocytic capacity. Consequently, this reduction in immune efficacy facilitates the progression of inflammatory diseases13. Senescent immune cells, especially macrophages, are considered particularly hazardous, primarily due to their capacity to accelerate organ aging, promote systemic aging, and demonstrate a heightened propensity for senescence compared to other cell types36. Senescent macrophages are pivotal in the secretion of proinflammatory mediators within tissues and the circulatory system, further exacerbating local and systemic chronic inflammation. This establishes a feedback loop that amplifies the accumulation of aging cells in the affected areas. Experiments conducted under controlled conditions demonstrated that macrophages exposed to LPS derived from P.g for 24 h exhibited significant increases in senescence markers, including p16, p21, and SA-β-Gal. This observation suggests that prolonged infection with gram-negative bacteria can induce DNA damage, leading to cellular senescence. Furthermore, our experimental results indicate that senescent macrophages secrete substantial amounts of inflammatory cytokines, including IL-6, IL-1β, and TNF-α, exacerbating tissue damage. For instance, IL-1β is crucial in initiating and progressing periodontal inflammation and bone degradation37. In periodontitis, macrophage-derived cytokines, including IL-1β, can activate osteoclasts and promote bone resorption38. Additionally, the persistent release of inflammatory mediators can establish a pro-senescent inflammatory microenvironment, thereby increasing the prevalence and diversity of senescent cells and perpetuating a deleterious cycle of cellular aging. Therefore, targeting the reduction or clearance of senescent macrophages may help regulate the local inflammatory microenvironment, essential for preventing further destruction of periodontal tissues and fostering conditions conducive to periodontal tissue regeneration. Our experimental findings indicate that inhibiting CSF1R expression can modulate inflammation and mitigate macrophage senescence.
The CSF1R, which belongs to the tyrosine-protein kinase receptor family, is responsible for regulating myeloid cell development, viability, and proliferation. CSF1R directly influences the differentiation and maturation of bone marrow mononuclear cells, culminating in the formation of macrophages and significantly contributing to innate immunity39. CSF1 and IL-34 are potent ligands for CSF1R, with IL-34 binding to CSF1R to initiate various signaling pathways, including the STAT and NF-κB pathways. These pathways significantly regulate cytokine expression40. Elevated CSF1 levels are detectable in the saliva and gingival tissues of patients with periodontitis and in the peri-implant environment. TNF-α and IL-1β, among other factors, can exacerbate periodontal inflammation by upregulating CSF1 expression through the CSF1R pathway41,42. In LPS-induced experimental periodontitis, CSF1R inhibition has been observed to confer bone-protective effects by inhibiting osteoclast formation and reducing the secretion of monocyte chemoattractant protein-1, which is highly expressed in periodontitis43. Our findings indicate that CSF1R expression is upregulated in periodontitis, and animal experimental results corroborate that inhibiting CSF1R expression can alleviate the disease. These findings align with those of Clark30 and Boström37. Therefore, targeting CSF1R has emerged as a promising therapeutic strategy for periodontitis because CSF1R inhibitors have demonstrated efficacy in mitigating periodontal inflammation by reducing macrophage senescence. Further research is needed to clarify the regulatory mechanisms of CSF1R inhibitors, which are crucial for developing effective clinical treatments.
Excessive generation of ROS, which results in oxidative stress-induced bone resorption, has garnered significant attention in the study of periodontitis-related bone degradation44. Research has shown that ROS overaccumulation predominantly affects periodontal tissue through two principal mechanisms: (1) ROS directly damages tissues by causing lipid peroxidation, DNA damage, protein impairment, and oxidation of essential enzymes, and (2) ROS acts as signaling molecules or inflammatory mediators, triggering pertinent signaling pathways that drive inflammation and tissue impairment45. Following the disruption of macrophage homeostasis, factors such as SASP and ROS contribute to a self-perpetuating environment that accelerates macrophage aging and promotes senescence13. Our in vitro studies demonstrated a significant increase in ROS levels and CSF1R expression in senescent macrophages, likely resulting from an inflammatory response triggered by elevated ROS, which activated the CSF1R signaling pathway. However, further empirical substantiation is required to validate this hypothesis. The preservation of redox balance in the human body and evaluation of ROS production are critically influenced by the enzymes superoxide dismutase (SOD) and catalase (CAT), which also function as oxidative stress biomarkers46. FOXO1 functions as a transcription factor within cells and plays a pivotal role in regulating various physiological and pathological processes by controlling the expression of numerous genes. Within the nucleus, FOXO1 facilitates the transcription of downstream target genes, thereby modulating the expression of CAT and SOD, whose coordinated activity effectively converts ROS into water (H2O), thus providing protection against oxidative stress47. AKT, which functions upstream of FOXO1, plays a significant role in this process. Activation of the PI3K/AKT pathway leads to FOXO1 phosphorylation by AKT within the nucleus, triggering its translocation to the cytoplasm and subsequent inactivation48. This process attenuates the regulatory capacity of FOXO1 over ROS production. Our investigation found elevated levels of p-PI3K, p-AKT, and p-FOXO1 in senescent macrophages, which obstructed FOXO1’s nuclear localization and impaired its transcriptional activity. Furthermore, our results indicated that CSF1R inhibition effectively diminished the levels of p-PI3K, p-AKT, and p-FOXO1, suggesting that CSF1R inhibitors may reduce the generation of senescent macrophages by mitigating ROS production through the PI3K/AKT/FOXO1 signaling pathway.
Based on these findings, the results of both the in vivo and in vitro experiments suggest that CSF1R inhibitors can effectively reduce senescent macrophage production, potentially alleviating periodontitis. This evidence supports our primary hypothesis. Further in vitro investigations into the underlying mechanisms revealed that the optimal concentration of CSF1R inhibitors is comparable to that of PI3K inhibitors in modulating the PI3K/AKT/FOXO1 pathway. Furthermore, PI3K inhibitors have been shown to attenuate macrophage senescence, suggesting that the effects of CSF1R inhibitors are mediated via this pathway. Elevated ROS levels in senescent macrophages likely activate the PI3K/AKT/FOXO1 signaling cascade, and CSF1R inhibitors appear to reduce ROS production through this mechanism. Further experiments are needed to provide direct evidence and confirm this hypothesis.
Moreover, This study has certain limitations that should be acknowledged. Firstly, the SASP is composed of a complex array of inflammatory cytokines, chemokines, and matrix-degrading enzymes. In this study, we primarily focused on the role of senescent macrophages as “inflammatory amplifiers” in periodontitis. As a result, we focused our analysis on a limited set of inflammatory cytokines, specifically IL-6, IL-1β and TNF-α, without extending to chemokines or matrix-degrading enzymes. Although this does not compromise the overall reliability of our findings, it remains a limitation of the study. Second, regarding the evaluation of cellular senescence, we assessed classical markers including p16, p21, and SA-β-gal. However, future studies should further investigate ultrastructural changes within senescent cells, such as alterations in organelle morphology and chromatin remodeling. Lastly, previous research has shown that senescent macrophages tend to exhibit a pro-inflammatory M1 phenotype49,50. Since the primary focus of our study was cellular senescence, we did not differentiate between the classical M1 and M2 macrophage phenotypes, which may limit the interpretation of macrophage functional states in our experimental model.
In summary, our research highlights a potential correlation between periodontitis progression and “two-source” inflammatory responses, with a particular focus on macrophage senescence. Additionally, our findings provide a theoretical framework for developing future targeted interventions aimed at senescent cells to alleviate inflammation-induced periodontal tissue destruction.
Conclusion
Our study demonstrated that CSF1R inhibitors can mitigate periodontitis in murine models by reducing senescent macrophage production. Additionally, the inhibitors attenuate macrophage senescence through the PI3K/AKT/FOXO1 signaling pathway. Our findings further suggest that the inhibition of CSF1R may decrease ROS expression by modulating the PI3K/AKT/FOXO1 signaling pathway, providing further evidence for its therapeutic potential in mitigating inflammation and tissue damage in periodontitis.
Data availability
Data will be made available on request from corresponding author.
References
Ortigara, G. B. et al. The 2018 EFP/AAP periodontitis case classification demonstrates high agreement with the 2012 CDC/AAP criteria. J. Clin. Periodontol.48(7), 886–895 (2021).
Genco, R. J. & Sanz, M. Clinical and public health implications of periodontal and systemic diseases: An overview. Periodontol. 200083(1), 7–13 (2020).
D′Aiuto, F. et al. Systemic effects of periodontitis treatment in patients with type 2 diabetes: a 12 month, singlecentre, investigatormasked, randomised trial. Lancet Diabetes Endocrinol. 6 (12), 954–965 (2018).
Potempa, J., Mydel, P. & Koziel, J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 13 (10), 606–620 (2017). (2017).
Van Dyke, T. E., Bartold, P. M. & Reynolds, E. C. The nexus between periodontal inflammation and dysbiosis. Front. Immunol. 11, 511 (2020).
Bartold, P. M. & Van Dyke, T. E. Periodontitis: a host-mediated disruption of microbial homeostasis. Unlearning learned concepts[J]. Periodontol. 203–217 (2013). (2000) 62(1).
Silva, N. et al. Host response mechanisms in periodontal diseases. J. Appl. Oral Sci.23, 329–355 (2015).
Aquino-Martinez, R. The emerging role of accelerated cellular senescence in periodontitis. J. Dent. Res.102(8), 854–862 (2023).
Chen, S. et al. Cellular senescence and periodontitis: Mechanisms and therapeutics. Biology11(10), 1419 (2022).
Aquino-Martinez, R. et al. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone132, 115220 (2020).
Ikegami, K. et al. Cellular senescence with SASP in periodontal ligament cells triggers inflammation in aging periodontal tissue. Aging (Albany NY). 15 (5), 1279 (2023).
Aquino-Martinez, R. et al. Senescent cells exacerbate chronic inflammation and contribute to periodontal disease progression in old mice. J. Periodontol. 92 (10), 1483–1495 (2021).
Nie, L. et al. lncRNA-triggered macrophage inflammaging deteriorates age-related diseases. Mediators Inflamm. 4260309 (2019). (2019)(1).
Prattichizzo, F. et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 15, 170–181 (2018).
Zhang, P. et al. Hyperglycemia-induced inflamm-aging accelerates gingival senescence via NLRC4 phosphorylation. J. Biol. Chem.294(49), 18807–18819 (2019).
Wang, Q. et al. Diabetes fuels periodontal lesions via GLUT1-driven macrophage inflammaging. Int. J. Oral Sci. 13 (1), 1–10 (2021).
Zhang, L. et al. Cellular senescence: A key therapeutic target in aging and diseases. J. Clin. Invest.132(15), e158450 (2022).
Roger, L., Tomas, F. & Gire, V. Mechanisms and regulation of cellular senescence. Int. J. Mol. Sci.22(23), 13173 (2021).
Martínez de Toda, I. et al. The role of immune cells in oxi-inflamm-aging. Cells10(11), 2974 (2021).
Ok, C. Y. et al. FK866 protects human dental pulp cells against oxidative stress-induced cellular senescence. Antioxidants 10 (2), 271 (2021).
Li, X. et al. Robust intervention for oxidative stress-induced injury in periodontitis via controllably released nanoparticles that regulate the ROS-PINK1-Parkin pathway. Front. Bioeng. Biotechnol.10, 1081977 (2022).
Kma, L. & Baruah, T. J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 69 (1), 248–264 (2022).
Kaneda, M. M. et al. PI3Kγ is a molecular switch that controls immune suppression. Nature539(7629), 437–442 (2016).
Rong, S. J. et al. The essential role of FoxO1 in the regulation of macrophage function. Biomed. Res. Int. 2022 (1), 1068962 (2022).
Muñoz-Garcia, J. et al. The twin cytokines interleukin-34 and CSF-1: Masterful conductors of macrophage homeostasis. Theranostics11(4), 1568 (2021).
Fujiwara, T. et al. CSF1/CSF1R signaling inhibitor pexidartinib (plx3397) reprograms tumorassociated macrophages and stimulates tcell infiltration in the sarcoma microenvironment. Mol. Cancer Ther.20(8), 1388–1399 (2021).
Butowski, N. et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy foundation early phase clinical trials consortium phase II study. Neuro Oncol.18(4), 557–564 (2015).
Smeester, B. A. et al. PLX3397 treatment inhibits constitutive CSF1R-induced oncogenic ERK signaling, reduces tumor growth, and metastatic burden in osteosarcoma. Bone136, 115353 (2020).
Giustini, N. et al. Tenosynovial giant cell tumor: case report of a patient effectively treated with pexidartinib (PLX3397) and review of the literature. Clin. Sarcoma Res. 8, 1–5 (2018).
Clark, D. et al. The contribution of macrophages in old mice to periodontal disease. J. Dent. Res.100(12), 1397–1404 (2021).
Xiao, T. J. et al. Regulation of colony-stimulating factor 1 receptor inhibitor pexidartinib on the senescence of mouse bone marrow-derived macrophages stimulated by lipopolysaccharide. Zhonghua Kou Qiang Yi Xue Za Zhi. 58 (6), 575–583 (2023).
Ashfaq, R. et al. Smart biomaterial gels for periodontal therapy: A novel approach. Biomed. Pharmacother. 183, 117836 (2025).
Xu, M. et al. Bupleurum chinense polysaccharide improves lps-induced senescence of RAW264.7 cells by regulating the NF-κB signaling pathway. Evid. Based Complement. Alternat. Med. 7060812 (2020). (2020).
Wang, H. et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging (Albany NY)12(10), 9240–9259 (2020).
Aquino-Martinez, R. et al. Periodontal disease and senescent cells: New players for an old oral health problem?. Int. J. Mol. Sci.21(20), 7441 (2020).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature594(7861), 100–105 (2021).
Boström, E. A. & Lundberg, P. The newly discovered cytokine IL-34 is expressed in gingival fibroblasts, shows enhanced expression by pro-inflammatory cytokines, and stimulates osteoclast differentiation. PLoS One8(12), e81665 (2013).
Sun, X. et al. Polarized macrophages in periodontitis: characteristics, function, and molecular signaling. Front. Immunol. 12, 763334 (2021).
Kumari, A., Silakari, O. & Singh, R. K. Recent advances in colony stimulating factor-1 receptor/cfms as an emerging target for various therapeutic implications. Biomed. Pharmacother.103, 662–679 (2018).
Truong, A. D. et al. Interleukin-34 regulates Th1 and Th17 cytokine production by activating multiple signaling pathways through CSF-1R in chicken cell lines. Int. J. Mol. Sci.19(6), 1665 (2018).
Clark, R. et al. Colony-stimulating factor‐1 receptor blockade attenuates inflammation in inflamed gingival tissue explants. J. Periodontal Res.56(6), 1141–1153 (2021).
Clark, R. et al. Expression of colonystimulating factor 1 and interleukin-34 in gingival tissue and gingival fibroblasts from periodontitis patients and controls. J. Periodontol.91(6), 828–835 (2020).
Wang, X. F. et al. Colonystimulating factor 1 receptor Inhibition prevents against lipopolysaccharideinduced osteoporosis by inhibiting osteoclast formation. Biomed. Pharmacother. 115, 108916 (2019).
Shi, L. et al. Crosstalk between reactive oxygen species and dynamin-related protein 1 in periodontitis. Free Radic. Biol. Med.172, 19–32 (2021).
Hirschfeld, J. et al. Modulation of neutrophil extracellular trap and reactive oxygen species release by periodontal bacteria. Infect. Immun. 85 (12), e00297–e00217 (2017).
Wang, J. et al. Molecular mechanism on cadmium-induced activity changes of catalase and superoxide dismutase. Int. J. Biol. Macromol. 77, 59–67 (2015).
Feng, B. et al. Li-ESWT treatment reduces inflammation, oxidative stress, and pain via the PI3K/AKT/FOXO1 pathway in autoimmune prostatitis rat models. Andrology9(5), 1593–1602 (2021).
Xu, S. et al. CD73 alleviates GSDMD-mediated microglia pyroptosis in spinal cord injury through PI3K/AKT/Foxo1 signaling. Clin. Transl Med. 11 (1), e269 (2021).
Chen, J. et al. Senescent macrophages trigger a pro-inflammatory program and promote the progression of rheumatoid arthritis. Int. Immunopharmacol. 149, 114164 (2025).
Covarrubias, A. J. et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab.2(11), 1265–1283 (2020).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant No. 82260193) and the Natural Science Foundation of Guizhou Province (Qiankehejichu - ZK[2024] General 251). Editorage (www.editage.cn) edited the English language for us.
Funding
National Natural Science Foundation of China, Grant/Award Numbers:82260193; Natural Science Foundation of Guizhou Province (Qiankehejichu - ZK[2024] General 251)
Author information
Authors and Affiliations
Contributions
J.K.: Research, data organization, and drafting the initial manuscript; J.Z.: Research, data organization, and revising the manuscript; Y.Y: Data acquisition, data evaluation and reviewing the manuscript; Y.W.: Data evaluation and writing support; T.X.: Experiment direction and writing support; L.L.: Data acquisition and writing support; H.F.: Data acquisition; A.T.: Funding acquisition, project management, reviewing and editing of the final manuscript. All authors have endorsed the final manuscript version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kang, J., Zhan, J., Yuan, Y. et al. CSF1R inhibitor (PLX3397) alleviates experimental periodontitis by reducing macrophage senescence through the PI3K/AKT/FOXO1 signaling pathway. Sci Rep 15, 27446 (2025). https://doi.org/10.1038/s41598-025-13153-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13153-6
Keywords
This article is cited by
-
Lactobacillus rhamnosus regulates airway epithelial cell senescence through the ADCK5/PI3K/AKT signaling axis and alleviates airway inflammation in asthma
Naunyn-Schmiedeberg's Archives of Pharmacology (2025)
