
Abstract
Newcastle disease virus (NDV), as an avian pathogen, can infect a broad spectrum of cell lines in vitro. However, noncarcinoma cell lines possessing nonsusceptibility to NDV are rare. Here, we isolated primary mouse embryonic fibroblasts (MEFs), which are nonsusceptible to NDV. MEF-derived cells were generated by passaging the cells over fifty times to achieve spontaneous immortalization. Two of the resulting cell lines were named SLM-21 and MEF50. Karyotype analysis revealed that SLM-21 has a near-tetraploid karyotype and that MEF50 shows a near-tetraploid and near-hexaploid chimeric karyotype. NDV exerted a significant cytopathic effect on MEF50, and substantial viral replication was observed. In contrast, NDV did not have a significant effect on SLM-21, indicating that SLM-21 was a nonsusceptible cell line to NDV, while MEF50 was a susceptible cell line. The NDV authentic sialic acid (SA) receptors SA 2,3-Gal and SA 2,6-Gal were expressed in SLM-21. Transcriptomic analysis revealed that the non-susceptibility of SLM-21 may be related to its broadly activated antiviral pathways and fine-tuned regulation of the cell cycle and DNA damage. This study provides a basic cell platform for exploring viral susceptibility and pathogenesis as well as host–virus interactions during NDV infection.
Similar content being viewed by others


Introduction
Newcastle disease virus (NDV) is a highly virulent avian-specific virus that poses a major threat to the global poultry industry1. NDV, also known as avian paramyxovirus serotype 1 (APMV-1) and Orthoavulavirus javaense (ICTV, 2022), is classified as a member of the genus Orthoavulavirus in the subfamily Avulavirinae under the family Paramyxovirinae2. According to its pathogenicity, NDV strains are categorized as lentogenic (low virulence), mesogenic (moderate virulence), or velogenic (highly virulent)3. Velogenic strains have a broad-spectrum ability to infect a wide range of bird species and present more pronounced neurological or gastrointestinal symptoms, ultimately leading to death4; however, they produce only limited clinical signs in nonavian hosts, such as conjunctivitis and elevated body temperature, and are not lethal to these animals5,6.
NDV can proliferate in diverse cell lines of multiple species7 including cell lines such as DF-1 (chicken)8 Vero (monkey)9 MDBK (dog)10 and L929 (mouse)11 and primary cells such as human primary dendritic cells12. NDV, a promising oncolytic agent, can extensively proliferate in various tumor cell lines, such as human fibrosarcoma (HT1080), cervical cancer (HeLa), liver cancer (HEpG2) and so on13,14. However, non-susceptible cells, which can almost completely block viral replication without significant CPE, are rarely found. Existing examples include Had-1 15 and Had-2 16 cell lines, which were derived from mouse mammary carcinoma FM3A cells through mutagenesis with N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) and showed resistance to NDV, which means reduced viral replication or slower progression of CPE. These lines provide valuable insights; however, their origin from carcinoma cells and induction by mutagenesis may introduce confounding factors when studying intrinsic host susceptibility in healthy cells. A human keratinocyte HaCaT17,18,19 and a human foreskin fibroblast LG1 20 as non-oncolytic control cell lines, were likely resistant to NDV replication in an NDV-oncolytic study. NDV selectively kills tumor cells in model mice without being cytotoxic to their somatic cells21,22,23,24,25 suggesting that mouse somatic cells may be nonsusceptible to NDV.
Normally, the ability of healthy cells to proliferate in vitro is strictly controlled. After a certain number of divisions, cells undergo replicative senescence and eventual death26. However, some cells, known as immortalized cells, have escaped the normal process of aging and cell death and acquired the ability to undergo continuous division. There are a few ways to immortalize cells with limited proliferation, such as physical and chemical stimulation, heterologous expression of viral oncogenes and restoration of telomerase activity27. Interestingly, increasing evidence indicates that some types of rodent cells, such as 3T3 fibroblasts and rat epidermal cells, can be immortalized spontaneously in vitro28,29 and the frequency of this immortalization is approximately 10−5 to 10−630. This spontaneous process is particularly advantageous as it allows for the establishment of immortalized cell lines without the addition of exogenous genes, thereby providing a more natural model for studying cell biology and host-pathogen interactions.
In this study, we aimed to establish stable, noncarcinoma mouse cell lines to serve as platforms for investigating NDV susceptibility and pathogenesis. We isolated primary mouse embryonic fibroblasts (MEFs) from BALB/c mice. MEFs were not susceptible to the highly virulent NDV strain F48E9. After over fifty continuous passages, we obtained two spontaneously immortalized cell lines, SLM-21 and MEF50. The two cell lines exhibited dramatic genetic differences in their karyotypes. SLM-21 was characterized as a nonsusceptible cell line to NDV, while MEF50 was a susceptible cell line. Furthermore, transcriptomic analysis of SLM-21 and MEF50 with/without NDV infection revealed that the differential cellular susceptibility to NDV is the result of its broad activation of antiviral innate immunity and fine-tuned regulation of the cell cycle and DNA damage. In conclusion, we established a platform for identifying noncarcinoma NDV-nonsusceptible cell lines and utilized it to obtain initial transcriptomic data and provide insights into the host natural selectivity of NDV.
Materials and methods
Viruses and cells
The NDV strains F48E9 and LaSota were purchased from the China Institute of Veterinary Drug Control. The F48E9 strain is a standard highly virulent virus that has been commonly used for challenge evaluation and vaccine development in China since 194831. The LaSota strain, an avirulent virus, is a globally used live vaccine in the poultry industry. The chicken/Jiangsu/17/2006 (JS/17) strain is a virulent virus and was kindly gifted by Dr. Hongjun Chen (Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences, China). rF48E9-eGFP is a recombinant F48E9 strain expressing an eGFP reporter that was established in our laboratory32. The viruses were propagated in 9- to 11-day-old SPF embryonated chicken eggs. Mouse embryonic fibroblast cell lines (NIH-3T3, ATCC CRL-1658), BHK-21 and Human immortalized keratinocytes cell line HaCaT (Sunncell, China) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin‒streptomycin (P/S).
Collection of organs and tissues
The mice were sacrificed by cervical dislocation and briefly rinsed in 70% (v/v) ethanol. The kidney and spleen were removed, washed in PBS and placed into a clean Petri dish. A pregnant mouse was sacrificed at 13 or 14 days postcoitum by cervical dislocation. Briefly, the sections were rinsed in 70% (v/v) ethanol. The uterine horns were dissected and placed into a Petri dish, and each embryo was separated from its placenta and embryonic sac. The head and organs were removed, the embryos were washed in PBS, and all the embryos were placed in a clean Petri dish.
Preparation of primary cells
The tissue was gently minced using a sterile razor blade until it became possible to pipette. The mixture was digested with 0.25% trypsin without EDTA (Gibco, Invitrogen) at room temperature for 15 min and stirred at low speed using a magnetic stirring apparatus. The cells were centrifuged at 1500 rpm for 5 min and suspended in PBS. The cells were centrifuged at 1500 rpm for 5 min, and DMEM supplemented with 10% FBS and 1% penicillin/streptomycin (P/S) was added. The cell resuspension solution was filtered through 149-, 74-, and 47-µm cell strainers and seeded in a cell culture dish.
Viral infection
Primary cells and cell lines were infected with the F48E9, JS/17 or LaSota strains at multiplicities of infection (MOIs) of 0.01, 0.1 or 1, respectively, at 37 °C for 1 h; then, the cells were washed three times with sterile PBS and cultured in DMEM containing 2% FBS. The culture medium was harvested at different hpi. The cytopathic effect (CPE) was visualized under an Olympus IX73 inverted research microscope (Japan). The viral titers of F48E9-infected MEF50 and SLM-21 cells were determined by a 50% tissue culture infective dose (TCID50/mL) assay, while those of LaSota-infected cells were determined by an immunofluorescence assay (IFA). The titers of F48E9-infected HaCaT cells were detected by plaque assay. The titer of TCID50/mL was calculated according to the method established by Reed and Muench.
Western blot analysis
All cell samples were lysed with 1× SDS‒PAGE loading buffer and then boiled for 10 min. After centrifugation, the supernatants were collected and subjected to 12% SDS‒PAGE. The proteins were transferred onto a polyvinylidene fluoride membrane (PVDF) (Millipore, United States). Immunoblotting was performed using the following primary antibodies: anti-NP rabbit pAb prepared by our laboratory and anti-β-actin mouse pAb (Proteintech, China). The secondary antibodies included goat anti-mouse IgG conjugated to horseradish peroxidase (HRP) (Proteintech, China) and goat anti-rabbit IgG (H&L)-HRP (Proteintech, China). An enhanced chemiluminescent (ECL) peroxidase substrate (Millipore) was used for the detection of proteins by using the Tanon 5200 Chemiluminescent Imaging System (Tanon, China).
Cell immortalization
Primary cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin‒streptomycin (P/S). When the cells had grown to more than 80% confluence, they were digested for 2 min using 0.25% trypsin and passed through a new 60 mm-diameter cell culture dish at a 1:3 ratio. After 50 passages, the cell morphology no longer changed, and the growth rate stabilized.
Cell growth kinetics
In brief, single cells were evenly seeded into 24-well plates at 4 × 104 cells per well to analyze growth for 1 to 5 days and the ½ medium was replaced with fresh medium/24 h. Cells were harvested at 24 h interval up to 5 days. A final volume of 1 ml brought up to for cell number count by hemocytometer. Trypan blue stained cells were considered as dead cells and non-stained cells were counted. Triplicate copies of cells were counted each time, and the mean cell counts were used to construct the growth curve.
Karyotype analysis
Chromosomes were prepared from SLM-21 or MEF50 at passage 83. The cells were exposed to 0.1 mg/mL colchicine in fresh medium and incubated at 37 °C. After 5–6 h, the cells were trypsinized and collected by centrifugation at 1000 rpm for 10 min. The cells were treated with hypotonic buffer containing 0.075 mol/L potassium chloride (KCl) in a 37 °C water bath for 32 min. The cells were fixed on precooled slides. Slides were stained with Giemsa solution in PBS for 10 min at RT. The Giemsa solution was gently rinsed with water, the slides were allowed to dry naturally, and then the cells microscopically examined.
Immunofluorescence assay
The cells were fixed with 4% paraformaldehyde in PBS for 15 min, washed three times with PBS, and permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature. After blocking with PBS containing 1% bovine serum albumin (BSA) for 1 h at 37 °C, the cells were incubated with the following primary antibodies or lectin at 4 °C overnight: (1) rabbit anti-NP pAb (1:100); (2) rabbit anti-vimentin mAb (1:100) (Zen-bioscience, China); (3) lectin-digoxigen in (DIG)-labeled Maackia amurensis lectin (MAA), specific for alpha-2,3-linked SA; and lectin-DIG-labeled Sambucus nigra agglutinin (SNA), specific for alpha-2,6-linked SA (Roche, Germany). After being washed with PBS three times, the samples were incubated with the secondary antibodies goat anti-rabbit IgG/fluorescein isothiocyanate (FITC) (Proteintech, China) or anti-digoxin-FITC (Sigma-Aldrich) for 30 min at 37 °C. Finally, the samples were counterstained with DAPI (Solarbio, China). The cells were observed and photographed under an Olympus IX73 inverted microscope.
Transcriptomic sequencing
SLM-21 and MEF50 cells were infected with or without rF48E9-GFP at an MOI of 5. Then, the cells were collected at 24 hpi (three biological replicates per group). All samples were then sent to Novogene (China) for transcriptome sequencing and analysis. The sequencing and analysis steps were as follows: Total RNA was extracted with TRIGene Reagent (Genestar, China) according to the manufacturer’s instructions. RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, United States).
Total RNA was used as input material for the RNA sample preparations. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using random hexamer primers and M-MuLV reverse transcriptase (RNase H-). Second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3’ ends of the DNA fragments, adaptors with hairpin loop structures were ligated to prepare for hybridization. To preferentially select cDNA fragments 370 ~ 420 bp in length, the library fragments were purified with an AMPure XP system (Beckman Coulter, United States). Then, PCR was performed with Phusion High-Fidelity DNA polymerase, universal PCR primers and Index (X) Primer. Finally, the PCR products were purified (AMPure XP system), and library quality was assessed on an Agilent Bioanalyzer 2100 system.
Quality control
Raw data (raw reads) in fastq format were first processed through in-house Perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing adapters, reads containing poly-N and low-quality reads from the raw data. At the same time, the Q20, Q30 and GC contents of the clean data were calculated. All downstream analyses were based on high-quality clean data.
Mapping reads to the reference genome
The reference genome and gene model annotation files were downloaded from https://ftp.ensembl.org/ directly. The index of the reference genome was built using HISAT2 v2.0.5, and paired-end clean reads were aligned to the reference genome using HISAT2 v2.0.5. We selected HISAT2 as the mapping tool because HISAT2 can generate a database of splice junctions based on a gene model annotation file and thus provides better mapping results than other nonsplice mapping tools.
Quantification of gene expression levels
FeatureCounts v1.5.0-p3 was used to count the number of reads mapped to each gene. Then, the FPKM of each gene was calculated based on the length of the gene and the number of reads mapped to the gene.
Differential expression analysis
Differential expression analysis was performed using the DESeq2 R package (1.20.0). DESeq2 provides statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with an adjusted P value < = 0.05 according to DESeq2 were considered to be differentially expressed.
GO and KEGG enrichment analysis of DEGs
Gene Ontology (GO) enrichment analysis of differentially expressed genes was implemented by the clusterProfiler R package, in which gene length bias was corrected. GO terms with corrected P values less than 0.05 were considered significantly enriched for DEGs. KEGG is a database resource for understanding high-level functions and utilities of biological systems, such as cells, organisms and ecosystems, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies33 (http://www.genome.jp/kegg/). We used the clusterProfiler R package to test the statistical enrichment of DEGs in KEGG pathways.
qRT-PCR
Total RNA from each group was reverse-transcribed with a StarScript II RT Kit (Genestar, China) according to the manufacturer’s instructions. The concentration of the synthesized cDNA was measured using a NanoDrop™ One Spectrophotometer (Thermo Fisher, United States), and the cDNA was stored at − 20 °C until analysis.
Four random genes were selected for confirmation, and the primers used for qRT-PCR assays are listed in Table 1. Real-time qPCR was performed on an Applied Biosystems QuantStudio 6 Flex real-time PCR system (Applied Biosystems) using 2x Fast qPCR Master Mixture (DiNing, China) according to the manufacturer’s instructions. All primer pairs were selected according to their specificity, as determined with dissociation curves. The PCR cycling conditions were as follows: one cycle at 94 °C for 2 min, 40 cycles of denaturation at 94 °C for 15 s and extension at 60 °C for 30 s, followed by a dissociation curve analysis step. Each sample was analyzed in triplicate.
The relative expression of the genes in the NDV group and in the mock group was calculated with the 2−∆∆Ct method and quantified relative to the housekeeping gene encoding beta-actin (β-actin), which was used as the endogenous control to normalize the expression levels of the genes and is expressed as the fold change in gene expression.
Statistical analysis
Two-tailed Student’s t test was used to estimate the statistical significance between two columns by GraphPad Prism 8. All data from three independent experiments are presented as the mean ± standard deviation (SD) of triplicate samples (n = 3); p < 0.05 was considered to indicate statistical significance. ns, not significant, ∗p < 0.05; ∗∗p < 0.01; ***p < 0.001.
Results
Susceptibility of different mouse primary cells to NDV
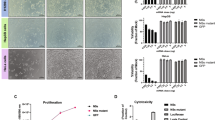
To verify the susceptibility of mouse primary cells to NDV, we isolated primary cells from the kidneys and spleens of BALB/c mice and the fibroblasts of a pregnant mouse embryo. These primary cells were infected with the highly virulent NDV strain F48E9 at an MOI of 1. Primary kidney cells showed CPEs, but there were no significant changes in MEFs or spleen primary cells (Fig. 1a). IFA analysis of the viral NP protein in infected cells revealed viral protein expression in kidney and spleen primary cells but not in MEFs (Fig. 1b). Furthermore, the Western blotting results also indicated the obvious expression of viral NP in kidney cells and decreased expression in spleen cells, but no viral NP was detected in MEFs (Fig. 1c, d). The viral titer in the culture supernatant showed that only kidney primary cells were permissive to NDV replication, while neither MEFs nor spleen primary cells showed significant viral replication (Fig. 1e). These results suggested that MEFs are nonsusceptible to NDV infection.

Susceptibility of different mouse primary cells to NDV. (a) Primary cells isolated from different tissues of mice were infected with the NDV F48E9 strain at an MOI of 1 and photographed 36 h after infection. The red arrow indicates syncytial formation (scale bar = 50 μm). (b) Immunofluorescence detection of NP protein in primary cells after infection with an MOI of 1 for 48 h (scale bar = 20 μm). (c) Western blot analysis of viral NP protein in primary cells infected with F48E9 (MOI = 1, 48 hpi). (d) The expression levels of viral proteins relative to that of β-actin were analyzed by densitometry. (e) Viral titers at 0, 24 and 48 hpi in primary cells. The data are representative of three independent experiments.
Immortalization of MEF cell line
A total of three independent MEF separations were performed to induce spontaneous immortalization, and each separation yielded cell lines containing four independently cultured clones. In initial passages, all cell lines were passaged every three to four days. In one separation, all four clones experienced massive cell death after each passage, and after five passages, the cell count could not be maintained. From the tenth to the forty-fifth generation, the proliferation rate of all cell lines gradually increased to once every one or two days and maintained a stable proliferation rate over the subsequent ten passages, with a spindle-shaped morphology similar to that of the primary cells. After passages, eight cell lines were obtained from twelve primary cell lines. Two of them, SLM-21 and MEF50, were evaluated for growth kinetics (Fig. 2a). The cell multiplication time was 11.22 h for NIH-3T3 cells, 15.90 h for SLM-21 cells, and 14.85 h for MEF50 cells. Normal mice have a diploid karyotype with 40 chromosomes. SLM-21 cells showed a stable near-tetraploid genome, with all twelve analyzed cells displaying chromosome counts between 71 and 76, indicating low chromosomal variability (Fig. 2b). Some chromosomal aberrations were present in the unpaired and mid-thickness chromosomes (Fig. 2c). In contrast, the MEF50 cell line exhibited a near-tetraploid and near-hexaploid chimeric karyotypes, with significant genomic instability: among fifteen analyzed cells, eleven had 64–79 chromosomes, while four cells ranged from 116 to 143 (Fig. 2b). suggesting that dramatic chromosomal changes were produced during MEF immortalization and that the changes were not nearly the same between these two cell lines.
To determine whether SLM-21 and MEF50 were fibroblasts, vimentin was used as a fibroblast marker for identification. Vimentin is an intermediate filament, indicating the mesenchymal origin of fibroblasts. Both cell lines were positively stained with an anti-vimentin antibody (Fig. 2d), suggesting that these cells were indeed fibroblasts.

Identification of immortalized cell lines. (a) Growth curves of SLM-21, MEF50 and NIH-3T3 cells. (b) Chromosome counts of SLM-21 and MEF50. (c) Typical karyotype analysis of SLM-21 and MEF50. (d) Immunofluorescence detection of SLM-21 and MEF50 with the fibroblast marker vimentin. (Scale bar = 20 μm.)
SLM-21 is nonsusceptible to NDV
To evaluate the susceptibility of SLM-21 and MEF50 to NDV, the cells were infected with F48E9-eGFP, which is a recombinant NDV strain capable of expressing green fluorescent protein upon cell infection. After infection with rF48E9-eGFP at an MOI of 1, we detected the expression of eGFP in NIH-3T3 and MEF50 cells (Fig. 3a), and Western blot analysis revealed the expression of the viral protein (Fig. 3b). However, the infected SLM-21 cells showed no fluorescence and no viral NP expression, and the virus also did not show significant replication in the infected cells (Fig. 3c), which was similar to the results in primary cells. In addition, when SLM-21 and NIH-3T3 cells were infected with the avirulent strain LaSota and the virulent strain JS/17, respectively, the proliferation of the JS/17 virus was significantly lower in the SLM-21 cells than in the NIH-3T3 cells, suggesting that SLM-21 cells are nonsusceptible to all genotypes of NDV (Fig. 3d). HaCaT was reported to be NDV-resistant cells as a non-oncolytic control in a previous oncolytic study18. Herein, we compared the nonsusceptibility to NDV between SLM-21 and HaCaT. Both cell lines were infected with rF48E9-eGFP at an MOI of 0.01. The number of observed fluorescence-positive cells increased along with post-infection time in the HaCaT (Fig. 3e). However, none of the fluorescence-positive cells were observed in the SLM-21. The rF48E9-eGFP was capable of proliferating well in the HaCaT, whereas it failed to replicate in the SLM-21 (Fig. 3f). Western blot data confirmed that the viral NP protein was expressed in the HaCaT, but not in the SLM-21 (Fig. 3e). These results indicated that the HaCaT allows limited NDV infection and replication, the SLM-21 cell line was not susceptible to NDV after immortalization of the primary cells and that MEF50 was susceptible to NDV.

The viral susceptibility of the immortalized cell lines SLM-21 and MEF50. (a) Viral infection and CPE in NIH-3T3, SLM-21 and MEF50 cells infected with F48E9-eGFP at an MOI of 1. (Scale bar = 50 μm). (b) Western blot analysis for detecting viral NP proteins. NIH-3T3 and SLM-21 cells were infected with F48E9, and SLM-21 and MEF50 cells were infected with F48E9-eGFP at an MOI of 1. (c) The viral replication of F48E9 at an MOI of 1 in NIH-3T3 and SLM-21 cells. (d) The viral replication of LaSota, JS/17 and F48E9 at an MOI of 1 at 24 hpi in NIH-3T3 and SLM-21 cells. (e) Viral infection and CPE in HaCaT and SLM-21 cells infected with F48E9-eGFP at an MOI of 0.01. (Scale bar = 200 μm). (f) Western blot analysis for detecting viral NP proteins. (g) The viral replication of F48E9-eGFP at an MOI of 0.01 in HaCaT and SLM-21 cells. *p < 0.05, **p < 0.01, and ***p < 0.001 (two-tailed unpaired Student’s t test). The data are representative of three independent experiments.
Sialic acid receptors were present on the surface of SLM-21
The host receptor is critical for viral susceptibility. To investigate whether SLM-21 lacks the NDV authentic sialic acid (SA) receptors SA 2,3-Gal and SA 2,6-Gal, SNA and MAA staining were performed on the surface of the cells. SLM-21 was positively stained by SNA and MAA but not by neuraminidase, a sialic acid hydrolyzer (Fig. 4a), indicating that the viral receptors SA 2,3-Gal and SA 2,6-Gal were present on the surface of SLM-21. Furthermore, the RNA expression of the viral NP gene was observed in SLM-21 after F48E9 strain infection (Fig. 4b). These results suggested that sialic acid receptors were not responsible for the nonsusceptibility of the virus to SLM-21.

The expression of viral SA receptors in SLM-21 cells. (a) SNA and MAA detected the cells with or without treatment with 2 mM neuraminidase for alpha-2,3-linked SA and alpha-2,6-linked SA, respectively. (b) The mRNA expression of viral NPs was measured by qRT‒PCR in NIH-3T3 and SLM-21 cells after infection with F48E9 at an MOI of 1. The data are representative of three independent experiments.
Transcriptomic analysis of SLM-21 and MEF50
To elucidate the mechanisms underlying the differential susceptibility of MEF50 and SLM-21 to NDV, transcriptomic analysis of both cell lines with/without infection was performed. A total of 46,180,068 raw reads were obtained. For the quality and reliability of the data analysis, the raw reads were strictly filtered, resulting in 44,727,841 clean reads. The number of unique mapped reads ranged from 34,855,331 to 44,641,474, approximately 79.89–91.46% of which were uniquely mapped to the reference genome (Table 2).
After quantifying gene expression levels, Pearson correlation coefficients were calculated for each pair of samples, and the results were analyzed using a heatmap. Each column of the heatmap represents the dependency of the X-axis parameter on the Y-axis parameter (Fig. S1). The gene expression patterns among biologically replicated samples were highly similar, indicating that the data could be used for the analysis of differentially expressed genes (DEGs).
To obtain a global view of the effects of NDV infection on both cell lines, the data of the four groups were compared. Overall, 906 DEGs were upregulated and 901 were downregulated in the NDV group compared with the mock group for SLM-21 cells. A total of 1411 DEGs were upregulated and 1448 were downregulated in the NDV group compared with the mock group for MEF50 cells. Among the DEGs in the mock group between the two cell lines, 1304 were upregulated and 1054 were downregulated, and among the DEGs in the NDV-infected group between the two cell lines, 1583 were upregulated and 1402 were downregulated (Fig. 5a). The list of all DEGs in each comparison group is displayed in additional file 1. The results indicate significant differences in DEGs between the two cell lines after immortalization.
To provide a more direct representation of the two cell lines, Venn analysis revealed 896 and 1948 DEGs between the infected SLM-21 and MEF50 strains, respectively (Fig. 5b). The results suggested that NDV infection had a broader impact on gene expression in the susceptible cell line MEF50 than in the nonsusceptible cell line SLM-21.

Transcriptomic analysis of the SLM-21 and MEF50 strains infected or mock-infected with the F48E9 strain. (a) Volcano plots. (b) Venn diagram of DEGs. (c) All significant GO terms and (d) the top 20 significant KEGG pathways of DEGs.
Enrichment analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were conducted on the DEGs of the NDV vs. mock groups in the SLM-21 and MEF50 groups to further compare the differences during NDV infection. Gene Ontology (GO) analysis can be subdivided into three categories: biological process (BP), cellular component (CC) and molecular function (MF). For the GO enrichment analysis, padj < 0.05 was used as the threshold for significant enrichment. Among the upregulated genes, the enriched BP terms in MEF50 were associated with RNA metabolism and regulation processes, protein translation and synthesis, cellular metabolism and energy metabolism, immune response and immune system regulation. The enriched MF terms were related to RNA-related functions, protein synthesis-related functions, enzyme activity, signal transduction-related functions, and transmembrane transport-related functions. For upregulated genes in SLM-21, the enriched BP terms were involved in DNA synthesis, replication, and metabolic processes, as well as immune system regulation and response processes. The enriched CC terms were associated with protein degradation and metabolic regulation. The enriched MF terms were also involved in signal transduction-related functions. These genes were also enriched in protein binding to nucleotides or nucleosides, catalytic activity for DNA synthesis and replication processes, and receptor regulation and activation activity.
For the downregulated genes, the enriched BP terms of MEF50 were mainly associated with cell adhesion processes, extracellular regions, and calcium ion binding processes. The enriched CC terms were mainly associated with the extracellular matrix, extracellular regions, and cell‒cell adhesion. The enriched MF terms were mainly associated with enzyme activity and regulation functions. These genes were also enriched in signal transduction and receptor functions. For downregulated genes in SLM-21, the enriched BP terms were also involved in cell adhesion processes and transmembrane transportation. The enriched CC terms were also associated with the extracellular matrix. These genes were also enriched in the cell membrane. The enriched MF terms were mainly associated with transmembrane transport. These genes were also enriched in enzyme activity, receptor activity, and signal transduction functions (Fig. 5c, Additional file 2). These results showed that immune-related processes were enriched in both cell lines, suggesting that the immune system of both cell lines is able to respond normally after viral infection.
Twenty-three upregulated and Thirty-three downregulated KEGG pathways were enriched in the MEF50, with 40 upregulated and 13 downregulated pathways enriched in SLM-21. The top 20 significantly enriched pathways were selected for plotting (Fig. 5d). A comprehensive list of the enriched pathways and genes can be found in Additional file 3. By comparison, we found SLM-21 was enriched in antiviral-related pathways, including influenza A, Epstein-Barr virus infection, NOD-like receptor signal pathway, etc. These pathways contain many key genes such as Stat1, Stat2, Irf7, Ifnb1, Ccl5, Cxcl10, Tlr3, Tlr7, Myd88, MDA5, and Oas1a, indicating that the SLM-21 cells induced a broad and potent antiviral response after NDV infection. Although MEF50 also enriched several antiviral pathways, its quantity and significance are both lower than SLM-21. Meanwhile, MEF50 enriched two pathways related to biosynthesis and growth: ribosome biogenesis in eukaryotes and aminoacyl-tRNA biosynthesis, which shows that the translation system of the host may be hijacked by the virus. Additionally, SLM-21 enriched several DNA damage response pathways, including homologous recombination and mismatch repair, none of which were enriched in MEF50. Interestingly, we found that the Fanconi anemia pathway was enriched only in SLM-21, whereas the expression of none of the MEF50-related genes changed after infection.
Verification of DEGs
To confirm the reliability of the transcriptomic data, qRT‒PCR was used to validate the DEGs between SLM-21 and MEF50 at 24 hpi (Fig. 6). The expression levels of eight selected genes were consistent with those of the transcriptomic profiles, indicating that the transcriptome data were reliable.

Verification of DEGs by qRT‒PCR in the SLM-21 and MEF50 cell lines infected or mock-infected with the F48E9 strain. The data are representative of three independent experiments.
Discussion
Cell lines are basic tools and platforms for virological research. Many cell lines are susceptible to NDV proliferation in vitro, even though they are derived from different species of birds and mammals, including humans7,12. However, less studies reported NDV-nonsusceptible cell lines. Our study aimed to address this gap by establishing a stable, noncarcinoma NDV-nonsusceptible cell line to serve as a valuable platform for elucidating host restriction factors. Using a spontaneous immortalization method, a nonsusceptible cell line, SLM-21, and a susceptible cell line, MEF50, derived from MEFs were obtained. The SLM-21 cell line showed nonsusceptible to viral proliferation and CPE. Furthermore, dramatic chromosome number abnormalities were detected in SLM-21. The transcriptomic profiles revealed that MEF50 but not SLM-21 was enriched in a large number of genes involved in protein synthesis. More virus-related pathways were enriched in SLM-21 than in MEF50. These differences suggest a role in the nonsusceptibility of SLM-21 to NDV.
NDV is an avian-specific pathogen. There is no strong evidence that NDV causes apparent diseases in other species, such as humans and other mammals34even if there are sporadic reports of the virus isolated from humans35,36,37. Mammals, such as mice, are generally considered to be nonsusceptible to NDV infection1,38. Therefore, mouse primary cells may theoretically be used to establish a nonsusceptible cell line for this virus. In fact, in our study, MEFs but not kidney or spleen primary cells were nonsusceptible to viral replication. A spontaneous immortalization method was utilized to establish cell lines. Spontaneous immortalization avoids the transduction of foreign genes, such as telomerase and viral antigens, which are used in most immortalization methods27. After the MEFs were continuously passaged for fifty generations, four cell lines were obtained. Among them, two cell lines, SLM-21 and MEF50, were identified as nonsusceptible and susceptible to NDV, respectively. The viral proliferation and CPE of NDV on MEF50 cells were similar to those on the commonly used cell line NIH-3T3, which is also derived from murine embryonic fibroblasts. However, these effects were not observed in SLM-21 cells. The susceptibility of cells to viruses is commonly related to viral receptors. The question is whether the nonsusceptible cell line SLM-21 lacks the sialic acid (SA) receptor of NDV. According to the SNA and MAA staining results, SA 2,3-Gal and SA 2,6-Gal were present on the surface of SLM-21, indicating that these viral receptors were not responsible for the nonsusceptibility of SLM-21 to NDV. On the other hand, low levels of viral mRNA expression were detected in the infected SLM-21 strain. On the other hand, while the virus can enter SLM-21 cells through SA receptors, only low levels of viral mRNA expression were detected, and no significant viral protein or progeny virus were produced. This strongly indicates that the non-susceptibility of SLM-21 primarily arises at a post-entry stage of the viral life cycle, where subsequent viral processes such as viral RNA transcription, genome replication, protein translation, virion assembly, or release are severely limited. This suggests the involvement of potent host intracellular restriction factors that act after viral entry.
Mice and MEF primary cells have diploid karyotypes with 40 chromosomes39. The karyotype of SLM-21 was aneuploid, nearing tetraploid, while the MEF50 exhibited near-tetraploid and near-hexaploid chimeric karyotypes. The genetics of both cell lines were intrinsically altered during spontaneous immortalization. Similarly, the karyotype of the NIH-3T3 cell line was near-tetraploid after the spontaneous immortalization of murine primary cells40. A human keratinocyte line was reported to be aneuploid after employing the same method17. Human vocal fold epithelial cell lines41 and swine umbilical vein endothelial cell lines42 immortalized by telomerase transduction methods also exhibit genomic mutations that cause chromosome changes. The genetic alteration of immortalized cells may result in the gain, loss or retention of the features of primary cells. In our study, SLM-21 retained its nonsusceptibility to NDV, similar to that of MEFs, whereas MEF50 lost these characteristics.
Previous studies reported that NDV-resistant cell lines, Had-115 and Had-216, were isolated from mouse carcinoma FM3A cells mutagenized with MNNG. Had-1 cell line lacks NDV-receptor and Had-2 secretes high levels of IFN during high-density culture, which results in their resistance to NDV. In noncarcinoma cells, HaCaT18,19 and LG1 20 were reported to be resistant to NDV as non-oncolytic control cells, while oncogenic Rac1 and N-Ras genes transduced or transfected HaCaT and LG1 were susceptible to NDV in oncolytic cytotoxicity. However, in the literature, HaCaT cells infected with NDV show viral protein expression and a significant increase in viral titers. This is similar to our results (Fig. 3e–g). Therefore, under our conditions, we believe that HaCaT can partly resist virus replication, but not be completely nonsusceptible. The LG1 showed similar aspects to HaCaT in the NDV oncolytic study20. We speculate that LG1 may also be susceptible to NDV, but it needs further investigation.
Transcriptome analysis revealed great differences between MEF50 and SLM-21. According to the GO enrichment analysis, the upregulated DEGs of MEF50 were enriched with a large number of genes related to protein synthesis, but those of SLM-21 were not enriched. According to the KEGG results, after NDV infection, SLM-21 upregulated multiple innate immune and antiviral response pathways, including “influenza A”, “NOD-like receptor signaling pathway“, “Toll-like receptor pathways” (Additional file 3), These pathways encompass pattern recognition receptors such as MDA5, TLR3, and TLR7, as well as signal transduction molecules including Myd88, Irf7, Stat1, and Stat2, leading to robust induction of the Ifnb1 gene and thereby initiating a broad antiviral state. Type I interferons induce the expression of hundreds of interferon-stimulated genes (ISGs), directly intervening in various stages of the viral life cycle, including replication, assembly, and budding, thereby effectively inhibiting viral replication43. In contrast, although MEF50 cells also showed some activation of immune pathways, the intensity and broadness of this activation were significantly lower than that of SLM-21. Another hypothesis is that NDV may be able to effectively evade or suppress the immune defenses of MEF50. The strong activation of biosynthetic pathways in MEF50 may be the result of the virus hijacking the cell to achieve replication. Viruses typically manipulate the host cell cycle to obtain resources such as nucleotides and replication machinery, thereby creating favorable conditions for their own replication44. For example, NDV infection can induce cell cycle arrest at the G0/G1 phase or preferentially infect cells in the S/G2 phase to promote viral replication45. Therefore, the non-susceptibility of SLM-21 cells to NDV may also be closely related to the fine regulation of the cell cycle and DNA damage response. KEGG analysis revealed that several pathways in SLM-21 cells, including “DNA replication”, “cell cycle”, “p53 signaling pathway”, and multiple DNA repair pathways (such as the “Fanconi anemia pathway”, “homologous recombination”, “mismatch repair”, etc), were significantly upregulated, revealing that SLM-21 may adopt an active strategy to control cell destiny. Interestingly, almost all of the upregulated genes in SLM-21 were enriched in the Fanconi anemia pathway; this enrichment was not found in MEF50. The inhibition of this pathway enhances the transgene levels of rAAV in human primary cells46 which may be a direction for future validation. These findings may provide valuable insight into the mechanisms underlying the nonsusceptibility of SLM-21 to NDV.
Conclusion
In this study, we developed a mouse cell line that is not susceptible to NDV infection, SLM-21 was established through spontaneous immortalization. SLM-21 had dramatic genetic alterations and gene expression differences compared with those of the susceptible cell line MEF50. The transcriptomic profiles suggested that the non-susceptibility of SLM-21 to NDV may be related to its broadly activated antiviral pathways and fine-tuned regulation of the cell cycle and DNA damage. This study provides a cell platform for exploring viral pathogenesis and host restriction factors in NDV.
Data availability
The datasets analyzed during the current study are available from the NCBI database (BioProject: PRJNA1188507).
References
Alexander, D. J. Newcastle disease and other avian paramyxoviruses. Rev. Sci. Tech. 19, 443–462. https://doi.org/10.20506/rst.19.2.1231 (2000).
Amarasinghe, G. K. et al. Taxonomy of the order mononegavirales: Update 2019. Arch. Virol. 164, 1967–1980. https://doi.org/10.1007/s00705-019-04247-4 (2019).
Ganar, K., Das, M., Sinha, S. & Kumar, S. Newcastle disease virus: Current status and our understanding. Virus Res. 184, 71–81. https://doi.org/10.1016/j.virusres.2014.02.016 (2014).
Kaleta, E. F. & Baldauf, C. in Newcastle Disease. 197–246 (eds Alexander, D. J.) (Springer, 1988).
Burnet, F. M. Human infection with the virus of newcastle disease of fowls. Medical J. Australia 2, 313 (1943).
Kuiken, T. et al. Pigeon paramyxovirus type 1 from a fatal human case induces pneumonia in experimentally infected cynomolgus macaques (Macaca fascicularis). Vet. Res. 48, 80. https://doi.org/10.1186/s13567-017-0486-6 (2017).
Saif, Y. Diseases of Poultry (Wiley, 2009).
Jang, J., Hong, S. H. & Kim, I. H. Validation of a real-time RT-PCR method to quantify Newcastle disease virus (NDV) titer and comparison with other quantifiable methods. J. Microbiol. Biotechnol. 21, 100–108. https://doi.org/10.4014/jmb.1006.06006 (2011).
Ravindra, P. V. et al. Newcastle disease virus-induced cytopathic effect in infected cells is caused by apoptosis. Virus Res. 141, 13–20. https://doi.org/10.1016/j.virusres.2008.12.008 (2009).
King, D. J. Newcastle disease virus passage in MDBK cells as an aid in detection of a virulent subpopulation. Avian Dis. 37, 961–969 (1993).
Ito, Y., Tsurudome, M., Yamada, A. & Hishiyama, M. Induction of cell fusion in Newcastle disease virus-infected L929 cells by anti-L929 cell antisera. J. Gen. Virol. 68 (Pt 5), 1261–1266. https://doi.org/10.1099/0022-1317-68-5-1261 (1987).
Leung, L. W. et al. Ebolavirus VP35 suppresses IFN production from conventional but not plasmacytoid dendritic cells. Immunol. Cell. Biol. 89, 792–802. https://doi.org/10.1038/icb.2010.169 (2011).
Reichard, K. W. et al. Newcastle disease virus selectively kills human tumor cells. J. Surg. Res. 52, 448–453. https://doi.org/10.1016/0022-4804(92)90310-v (1992).
Elankumaran, S., Rockemann, D. & Samal, S. K. Newcastle disease virus exerts oncolysis by both intrinsic and extrinsic caspase-dependent pathways of cell death. J. Virol. 80, 7522–7534. https://doi.org/10.1128/jvi.00241-06 (2006).
Hara, T., Hattori, S. & Kawakita, M. Isolation and characterization of mouse FM3A cell mutants which are devoid of Newcastle disease virus receptors. J. Virol. 63, 182–188. https://doi.org/10.1128/jvi.63.1.182-188.1989 (1989).
Aoki, K., Oh-hira, M., Hoshino, M. & Kawakita, M. Isolation and characterization of a novel mutant mouse cell line resistant to Newcastle disease virus: Constitutive interferon production and enhanced interferon sensitivity. Arch. Virol. 139, 337–350. https://doi.org/10.1007/bf01310796 (1994).
Boukamp, P. et al. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell. Biol. 106, 761–771. https://doi.org/10.1083/jcb.106.3.761 (1988).
Puhlmann, J., Puehler, F., Mumberg, D., Boukamp, P. & Beier, R. Rac1 is required for oncolytic NDV replication in human cancer cells and establishes a link between tumorigenesis and sensitivity to oncolytic virus. Oncogene 29, 2205–2216. https://doi.org/10.1038/onc.2009.507 (2010).
Pühler, F. et al. Generation of a recombinant oncolytic Newcastle disease virus and expression of a full IgG antibody from two transgenes. Gene Ther. 15, 371–383. https://doi.org/10.1038/sj.gt.3303095 (2008).
Lorence, R. M. et al. Complete regression of human fibrosarcoma xenografts after local Newcastle disease virus therapy. Cancer Res. 54, 6017–6021 (1994).
de Graaf, J. F. et al. Comparison between intratumoral and intravenously administered oncolytic virus therapy with Newcastle disease virus in a xenograft murine model for pancreatic adenocarcinoma. Heliyon 8, e09915. https://doi.org/10.1016/j.heliyon.2022.e09915 (2022).
Chen, L. et al. Oncolytic activity of wild-type Newcastle disease virus HK84 against hepatocellular carcinoma associated with activation of type I interferon signaling. J. Clin. Transl Hepatol. 10, 284–296. https://doi.org/10.14218/jcth.2021.00284 (2022).
Syed Najmuddin, S. U. F. et al. Oncolytic effects of the recombinant Newcastle disease virus, rAF-IL12, against colon cancer cells in vitro and in tumor-challenged NCr-Foxn1nu nude mice. PeerJ 8, e9761. https://doi.org/10.7717/peerj.9761 (2020).
Yurchenko, K. S. et al. Oncolytic effect of wild-type Newcastle disease virus isolates in cancer cell lines in vitro and in vivo on xenograft model. PLoS One. 13, e0195425. https://doi.org/10.1371/journal.pone.0195425 (2018).
Al-Shammari, A. M., Jalill, R. D. A. & Hussein, M. F. Combined therapy of oncolytic Newcastle disease virus and rhizomes extract of rheum Ribes enhances cancer virotherapy in vitro and in vivo. Mol. Biol. Rep. 47, 1691–1702. https://doi.org/10.1007/s11033-020-05259-z (2020).
Zhao, X. et al. Spontaneous immortalization of mouse liver sinusoidal endothelial cells. Int. J. Mol. Med. 35, 617–624. https://doi.org/10.3892/ijmm.2015.2067 (2015).
Guo, D., Zhang, L., Wang, X., Zheng, J. & Lin, S. Establishment methods and research progress of livestock and poultry immortalized cell lines: A review. Front. Vet. Sci. 9, 956357. https://doi.org/10.3389/fvets.2022.956357 (2022).
Phillips, M. A. & Rice, R. H. Convergent differentiation in cultured rat cells from nonkeratinized epithelia: Keratinocyte character and intrinsic differences. J. Cell. Biol. 97, 686–691. https://doi.org/10.1083/jcb.97.3.686 (1983).
Todaro, G. J. & Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell. Biol. 17, 299–313. https://doi.org/10.1083/jcb.17.2.299 (1963).
Katakura, Y., Alam, S. & Shirahata, S. Immortalization by gene transfection. Methods Cell. Biol. 57, 69–91. https://doi.org/10.1016/s0091-679x(08)61573-3 (1998).
Guo, Y. et al. α2,3- and α2,6-linked Sialic acids are important for cell binding and replication of Newcastle disease virus in chicken primary neuronal cells. Acta Virol. 62, 235–245. https://doi.org/10.4149/av_2018_217 (2018).
Wei, Q. et al. The W195 residue of the Newcastle disease virus V protein is critical for multiple aspects of viral self-regulation through interactions between V and nucleoproteins. Viruses 16, 584 (2024).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Freeman, A. I. et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 13, 221–228. https://doi.org/10.1016/j.ymthe.2005.08.016 (2006).
Goebel, S. J. et al. Isolation of avian paramyxovirus 1 from a patient with a lethal case of pneumonia. J. Virol. 81, 12709–12714. https://doi.org/10.1128/jvi.01406-07 (2007).
Shabbir, M. et al. Genomic characterization of velogenic avian orthoavulavirus 1 isolates from poultry workers: Implications to emergence and its zoonotic potential towards public health. Asian Pac. J. Trop. Med. 14, 64–72. https://doi.org/10.4103/1995-7645.306762 (2021).
Ul-Rahman, A., Ishaq, H. M., Raza, M. A. & Shabbir, M. Z. Zoonotic potential of Newcastle disease virus: Old and novel perspectives related to public health. Rev. Med. Virol. 32, e2246. https://doi.org/10.1002/rmv.2246 (2022).
Rtishchev, A., Treshchalina, A., Shustova, E., Boravleva, E. & Gambaryan, A. An outbreak of Newcastle disease virus in the Moscow region in the summer of 2022. Vet. Sci. https://doi.org/10.3390/vetsci10060404 (2023).
Suckow, M. A., Hashway, S. & Pritchett-Corning, K. R. The Laboratory Mouse (CRC, 2023).
Aaronson, S. A. & Todaro, G. J. Development of 3T3-like lines from balb/c mouse embryo cultures: Transformation susceptibility to SV40. J. Cell. Physiol. 72, 141–148. https://doi.org/10.1002/jcp.1040720208 (1968).
Chen, X. et al. Novel immortalized human vocal fold epithelial cell line: In vitro tool for mucosal biology. Faseb J. 35, e21243. https://doi.org/10.1096/fj.202001423R (2021).
Hong, H. X., Zhang, Y. M., Xu, H., Su, Z. Y. & Sun, P. Immortalization of swine umbilical vein endothelial cells with human telomerase reverse transcriptase. Mol. Cells. 24, 358–363 (2007).
Luan, X., Wang, L., Song, G. & Zhou, W. Innate immune responses to RNA: Sensing and signaling. Front. Immunol. https://doi.org/10.3389/fimmu.2024.1287940 (2024).
Bagga, S. & Bouchard, M. J. Cell cycle regulation during viral infection. Methods Mol. Biol. 1170, 165–227. https://doi.org/10.1007/978-1-4939-0888-2_10 (2014).
Wang, Y. et al. Newcastle disease virus induces G(0)/G(1) cell cycle arrest in asynchronously growing cells. Virology 520, 67–74. https://doi.org/10.1016/j.virol.2018.05.005 (2018).
Ngo, A. M. & Puschnik, A. S. Genome-scale analysis of cellular restriction factors that inhibit transgene expression from adeno-associated virus vectors. J. Virol. 97, e0194822. https://doi.org/10.1128/jvi.01948-22 (2023).
Funding
This work was supported by the National Natural Science Foundation of China (31972662).
Author information
Authors and Affiliations
Contributions
S.X. proposed the concept and designed the methodology; H.L., Z.Z., C.C., J.L.and Q.W. proformed the experiment; H.L., Y.L., F.M., and J.T. analysed the data; H.L. and Z.Z. wrote the original draft; W.S. and Q.H. revised the manuscript; S.X. and Z.Y. administered the project; H.L. (Haijin Liu) supervised the study; S.X. acquired funding. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Nine-day-old SPF embryonated chicken eggs were purchased from Shandong HaoTai Co., Ltd. (China). Four-week-old SPF BALB/c mice were purchased from Chengdu Dossy Co., Ltd. (China) and fed SPF mouse maintenance feed and sterile distilled water in other cages. Bleeding of the mice was performed under anesthesia, which was induced by ketamine and xylazine. All efforts were made to minimize suffering. All animals were euthanized by using carbon dioxide. All animal experiments were approved by the Animal Care and Use Committee of Northwest A&F University, China, and were conducted in accordance with guidelines established by the Chinese Committee for Animal Experiments (approval number: 2021022). This study adhered to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, H., Zhou, Z., Chang, C. et al. A noncarcinoma mouse cell line is nonsusceptible to Newcastle disease virus established by spontaneous immortalization. Sci Rep 15, 29744 (2025). https://doi.org/10.1038/s41598-025-14404-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14404-2
