
Abstract
This study aimed at targeting hepatic cancer stem cells (CSCs) with quercetin (Q) or kaempferol (K) loaded into poly(lactide-co-glycolide) (PLGA) nanoparticles (NPs) decorated with CD133 antibody. For this purpose, the formulated Q NPs and K NPs and their free forms were evaluated for their cytotoxic potential, apoptotic activity, and anti-migratory effect against CD133+ CSCs isolated from the Huh7 cell line. Moreover, their influence on the hepatic CSCs-relevant molecular pathways was evaluated through analyzing several related gene expression levels. Interestingly, the in vitro study revealed that the Q NPs and K NPs and their free forms exhibit significant cytotoxic potential against CSCs isolated from the Huh7 cell line. The flow cytometric analysis revealed that Q NPs recorded the highest induction of apoptosis (77.8%) relative to the control (1.8%). The migration of hepatic CSCs is restrained by treatment with the suggested NPs and their free forms, but the most pronounced effect was observed after treatment with Q NPs. Both Q NPs and K NPs triggered significant down-regulation in the expression level of ABCG2, survivin, vimentin, cyclin D1, c-Myc, MMP-7, and VEGF genes in hepatic CSCs. The treatment with Q NPs motivated significant up-regulation in the expression level of the P53 gene in hepatic CSCs. Conclusively, the obtained results shed light on the success of Q NPs and K NPs modified with CD133 antibody on their surfaces in targeting hepatic CSCs. This effect was evidenced by their ability to significantly induce apoptosis, inhibit metastasis, reverse drug resistance, and interfere with CSC-associated signaling pathways.
Similar content being viewed by others


Introduction
Liver cancer is the 3rd leading cause of cancer-related deaths globally, with hepatocellular carcinoma (HCC) accounting for the majority of cases1. The incidence of HCC is rising by 2–4% annually across the global population and is projected to surpass one million cases per year by 20252. Similarly, Egypt has witnessed a nearly twofold increase in HCC cases over the last decade3. In Egypt, HCC is the primary cause of cancer-related morbidity and mortality and is the most prevalent cancer among males4. The average age of HCC onset in Egypt is 58 years, compared to 46 years in other countries5.
Surgery is the therapeutic choice for a limited number of patients with HCC, while the majority undergo locoregional treatments, including percutaneous ethanol injection, radiofrequency ablation, transarterial chemoembolization (TACE), radioembolization, radiotherapy, or transarterial radioembolization. Systemic treatment is typically reserved for patients who are ineligible for locoregional therapies or do not respond to TACE. Recently, the introduction of novel immunotherapies, such as immune checkpoint inhibitors, has started to transform the landscape of systemic HCC therapy6.
For years, liver cancer stem cells (CSCs) have been extensively studied as a key factor contributing to poor treatment outcomes and tumor relapse in liver cancer patients. CSCs, also recognized as tumor-initiating cells (TICs), constitute malignant cells within the tumor that retain self-renewal and differentiation capabilities, enabling them to drive tumorigenesis. These cells play a crucial role in therapeutic resistance, as they exhibit high resistance to conventional chemotherapy, evade apoptosis, survive treatment, and repopulate the tumor with limited proliferative potential. Additionally, CSCs can metastasize, leading to the formation of new tumors7.
Xenotransplantation and fluorescence-activated cell sorting (FACS) experiments have identified key indicators for CSCs in HCC, which can be categorized into intracellular indicators, like cytokeratin 19 (CK19), and surface indicators, such as CD13, CD24, CD44, CD47, CD90, CD133, and epithelial cellular adhesion molecule (EpCAM)8. Among these, CD133 is a widely recognized transmembrane glycoprotein and a universal CSC indicator over numerous cancers9. In HCC, CD133 is highly expressed in CSCs10 and has been recognized as a crucial target for enhancing the effectiveness of chemotherapy in recurrent HCC cases11. CD133-expressing HCC stem cells are also implicated in liver tumorigenicity and resistance to radiotherapy, primarily due to the activation of key signaling pathways, including AKT/Protein kinase B (PKB), B-cell lymphoma 2 (Bcl-2)12, and mitogen-activated protein kinase (MAPK)/PI3K13. Additionally, CSCs and normal stem cells engage many essential stemness-related signaling pathways, like Wnt/β-Catenin, Notch, nuclear factor kappa B (NF-κB), Hedgehog (HH), and Janus kinase/signal transducer and activator (JAK/STAT) transcription proteins in HCC14. Therefore, monitoring CSCs’ progression and regression is crucial for improving the diagnosis and treatment of HCC15.
Nanotechnology offers a promising and innovative approach for combating cancer, especially in targeting the evasive and treatment-resistant CSCs16. There is growing interest in utilizing nanoformulation-based anticancer medications, where NPs are particularly designed to recognize and target CSCs based on their molecular markers17. The NPs exhibit superior encapsulation capabilities, lack heterogeneity in tumors, and leverage the enhanced permeability and retention (EPR) effect, allowing for better accumulation within tumor regions. As a result, NP-based targeted therapy enhances therapeutic efficacy while reducing off-target effects18. NP delivery systems can be classified into passive and active targeting strategies. Passive targeting relies on the natural accumulation of NPs in tumor sites through the EPR effect19, but its effectiveness is limited due to tumor heterogeneity and insufficient accumulation20. To overcome these challenges, active targeting enhances the EPR effect and improves drug delivery efficiency beyond passive targeting alone21. This targeting is achieved by functionalizing the NP surface with ligands that facilitate affinity recognition, retention, and uptake by target cells22. This approach relies on specific intermolecular recognition23, where ligand-receptor interactions on target cell surfaces increase NP uptake, ultimately enhancing therapeutic efficacy24.
Polymeric nanoparticles are widely regarded as the most effective drug delivery vehicles due to their exceptional pharmacokinetic properties. They offer several advantages over conventional drug delivery systems, including high durability, compatibility with both hydrophobic and hydrophilic substances, strong resistance to degradation by enzymes, reduced frequency of drug administration, and, most importantly, minimized harmful side effects25. To specifically target and eliminate CSCs, CSC-specific antibodies or ligands can be conjugated onto the surface or interior of NPs26. A notable advancement in this field is the evolution of CD133-targeted NPs, developed to selectively deliver therapeutic candidates to cancer cells expressing the CD133 antigen. This targeting is fulfilled by modifying NP surfaces with antibodies, peptides, or small molecules that exhibit high affinity for CD133, ensuring precise recognition of CD133-expressing CSCs27. Studies have demonstrated that NPs functionalized with anti-CD133 antibodies and loaded with chemotherapeutic agents can effectively target CD133-positive CSCs, leading to tumor outgrowth inhibition in the liver cancer and glioblastoma mouse models28.
Natural compounds have long served as a valuable source of biologically active molecules capable of interacting with numerous cellular targets while minimizing the adverse side events usually linked with cancer treatments29. One such compound is quercetin (Q), a flavonoid with the chemical structure 3,3’,4’,5,7-pentahydroxyflavone30. It exhibits a broad range of biological functions, making it a hopeful candidate for cancer therapy. Its mode of action involves tumor cell cycle suppression, apoptosis induction, ROS modulation, and the reduction of chemotherapeutic resistance. Given these activities, pharmaceutical engineering has focused on developing advanced nanocarriers to enhance quercetin’s bioavailability and targeting capabilities, thereby improving its therapeutic efficiency against HCC and addressing anticancer drug resistance31. Ren et al.32 successfully synthesized Q NPs by utilizing gold NPs, Q solution, and poly (DL-lactide-co-glycolide) (PLGA) for liver cancer treatment. These Q NPs effectively inhibited cell growth of liver cancer and colony formation by upregulating p27 and downregulating c-Myc, cyclinD1, cyclin-dependent kinase 1 (CDK1), matrix metallopeptidase 7, and β-catenin within the cancer cells. These findings suggest that the Q NPs have significant potential as a multifunctional candidate for liver cancer therapy.
Kaempferol (K) (3,5,7-trihydroxy-2-(4-hydroxyphenyl)-4 H-chromen-4-one) is a flavonoid abundantly present in fruits and vegetables33, including onions, green tea, grapes, potatoes, tomatoes, apples, cucumbers, broccoli, blackberries, and green beans34. It functions as a free radical scavenger and supports the activity of several antioxidant enzymes. K exerts its chemopreventive effects through multiple mechanisms, including cell cycle arrest, anti-proliferation, anti-angiogenesis, and apoptosis induction35. Despite its extensive pharmacological properties, the biomedical application of K is limited due to its poor water solubility, low permeability, instability in alkaline aqueous environments, extensive metabolic processing before systemic absorption, and poor oral bioavailability36. A promising approach to overcoming these limitations is the evolution of NPs-based drug delivery systems, which enhance the oral bioavailability of hydrophobic and lipophilic compounds like K37 due to their high biocompatibility38. Mechanistic studies have demonstrated that K-conjugated NPs promote oxidative stress-mediated apoptosis and cell cycle arrest in liver cancer cells, highlighting their potential as an anticancer therapy33. For instance, K-coated silver NPs displayed a synergistic apoptotic effect in liver cancer (HepG2) cells. This was characterized by a reduction in B-cell leukemia/lymphoma 2 (Bcl-2) protein levels, an increase in Bcl-2-associated protein x (Bax) and cytochrome C levels, caspase-3 activation owing to mitochondrial membrane disruption, and enhanced p53-mediated cell cycle arrest. These findings underscore the potential of K-based nanotherapeutics in liver cancer treatment39.
Therefore, utilizing the unique advantages of nanotechnology for enhancing the therapeutic efficacy of HCC heralds a new era of precision medicine. Thus, this research underscores the potential of NPs-based technology in improving HCC treatment outcomes with special emphasis on the role of CD133-antibody conjugated PLGA NPs to selectively target and deliver Q or K to CSCs in the Huh7 cell line. The successful treatment of CSCs via influencing numerous cellular mechanisms simultaneously would prohibit the survival of this evasive subpopulation and consequently hepatic cancer metastasis, relapse, and drug resistance.
Materials and methods
Materials
Quercetin (Q), kaempferol (K), dichloromethane (DCM), poly(lactide-co-glycolide) (PLGA), polyvinyl alcohol (PVA), bovine serum albumin (BSA), 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich (USA). Regenerated cellulose membranes (Amicon 10,000 MWCO ultrafilter) and dialysis bag (MW cut-off 10 kDa) were supplied from Millipore (USA). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin, and phosphate-buffered saline (PBS) were procured from Biowest (France). CD133 MicroBeads kit, LS MACS column, phycoerythrin (PE)-conjugated CD24 and CD133 antibodies, as well as fluorescein isothiocyanate (FITC)-conjugated CD34 and CD326 antibodies, were obtained from Miltenyi Biotec. (Germany). Additionally, PE-conjugated CD44 and CD90 antibodies were purchased from R&D Systems, Abingdon (UK).
Methods
I. Nanoparticles Preparation and characterization
Preparation of nanoparticles
Q- and K-loaded PLGA nanoparticles (non-targeted) were prepared using the emulsification-solvent evaporation method, with slight modifications based on previously reported protocols40. Briefly, 10 mg of Q (quercetin) and 10 mg of K (kaempferol) were dissolved in 5 mL of dichloromethane (DCM) containing 100 mg of PLGA (50:50, Mw 30,000 Da), yielding a drug-to-polymer ratio of 1:10 (w/w). This organic phase was slowly added dropwise into 20 mL of an aqueous solution of 1% (w/v) polyvinyl alcohol (PVA; 89% hydrolyzed, Mw 30,000 Da) under continuous stirring. The resulting mixture was sonicated using a probe sonicator (e.g., Vibra-Cell VCX130, Sonics & Materials Inc., USA) at 40% amplitude for 2 min in an ice bath to form a stable oil-in-water (O/W) emulsion. To evaporate the organic solvent (DCM), the emulsion was stirred at 500 rpm at room temperature for 4 h on a magnetic stirrer. Following solvent evaporation, nanoparticles were recovered by centrifugation at 20,000 rpm for 30 min at 4 °C (using a high-speed centrifuge such as Beckman Coulter Avanti J-26 XPI). The pellets were washed three times with distilled water to remove residual PVA and unencapsulated drug. The purified nanoparticles were then lyophilized (Labconco FreeZone 2.5) and stored at 4 °C in airtight containers for further analysis.
Decoration of nanoparticles with CD133 antibody (targeted)
For active targeting, CD133 antibodies were covalently conjugated to the surface of PLGA nanoparticles using EDC/NHS chemistry. Lyophilized nanoparticles were first dispersed in 10 mL of PBS (pH 7.4), followed by the addition of 10 mg of EDC and 5 mg of NHS. The mixture was stirred at room temperature for 4 h to activate the carboxyl groups on the PLGA surface. Activated nanoparticles were then separated by ultracentrifugation at 25,000 rpm for 30 min at 4 °C to remove unreacted EDC and NHS. The pellet was resuspended in 5 mL PBS, and 50 µg of CD133 antibody was added. The conjugation mixture was stirred for 2 h at room temperature, followed by overnight incubation at 4 °C. On the following day, ultracentrifugation was repeated at 25,000 rpm for 30 min at 4 °C to remove unconjugated antibodies. The supernatant was collected, and unconjugated antibody was quantified using the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. The pellet (antibody-conjugated NPs) was lyophilized and stored at 4 °C. For control experiments, bovine serum albumin (BSA)-decorated nanoparticles were synthesized using the same conjugation protocol, replacing the CD133 antibody with BSA40.
Nanoparticles characterization
- Transmission electron microscope (TEM).
The morphology of the NPs was analyzed using TEM (JEOL JEM-1400). A 100 µg/mL suspension of the NPs was deposited onto Formvar-coated copper grids and allowed to dry completely at room temperature. Once dried, imaging and structural analysis were performed using Digital Micrograph and Soft Imaging Viewer Software, enabling detailed visualization of the NP size, shape, and surface characteristics.
- Particle size distribution and zeta potential.
The dynamic particle size and surface charge (zeta potential) of the prepared NPs were determined using a Dynamic Light Scattering (DLS) instrument (Zetasizer Nano ZS, Malvern Instruments, UK) equipped with a 633 nm laser. To ensure instrument accuracy, a reference standard (DTS1230, zeta-potential standard from Malvern) was used for calibration. For measurements, 1 mL of the NP suspension was placed in a disposable transparent sizing cuvette. Data analysis was performed using Malvern Instruments’ Dispersion Technology Software (Version 4.0). The values of zeta-potential were calculated from the measured electrophoretic mobility data using the Smoluchowski equation.
-
Fourier transform infrared spectroscopy (FTIR).
The functional groups of the developed NPs were analyzed using FTIR spectroscopy (Bruker Vertex 80v). The spectra were recorded over a wavelength range of 4000–400 cm⁻¹ with high resolution to identify characteristic functional group vibrations and confirm successful NP formulation and surface modifications.
-
Entrapment efficiency measurement.
The entrapment efficiency (EE%) of the NPs was detected by calculating the ratio of the incorporated Q and K to the total amount initially added during formulation according to Eq. (1). To remove impurities and free, non-conjugated compounds, the prepared nanoformulations underwent purification using the dialysis tubing technique. This process involved elution through regenerated cellulose membranes (Amicon 10,000 MWCO ultrafilter). The quantification of encapsulated compounds was performed using a microplate reader (BMG Labtech, Germany), with all data processed against calibration curves specific to each compound.
- Drug release profiles.
To evaluate the drug release profile, a specific amount of each nanoformulation and its corresponding free form were diffused in a freshly prepared release medium inside a dialysis bag (MW cut-off 10 kDa). The sealed dialysis bag was then immersed in 30 mL of release medium (pH 7.4) and placed in a shaking incubator at 37 °C under mild stirring to simulate physiological conditions. At predetermined time intervals (0, 1, 2, 3, 4, 5, and 24 h), 1 mL of the release medium was withdrawn and replaced with an equal volume of fresh medium to maintain sink conditions. The collected samples were filtered, and the concentration of the released drug was quantified using a microplate reader (BMG Labtech, Germany)41,42. Furthermore, drug release kinetic analysis was performed using mathematical relations43,44.
II. Cancer stem cells isolation and characterization
Cell line
The Huh7 cell line was purchased from VACSERA Co. (Egypt), which obtained it from the American Type Culture Collection Center. The cells were cultured in DMEM supplemented with 10% FBS and 100 IU/mL penicillin G − 100 µg/mL streptomycin. The cells were maintained at 37 °C in a humidified incubator with 5% CO₂ to ensure optimal growth conditions.
Isolation and culture of hepatic cancer stem cells (CSCs)
CD133⁺ hepatic CSCs were isolated from the Huh7 cell line using the CD133 MicroBeads kit following the manufacturer’s protocol. The cells were magnetically labeled with CD133 MicroBeads – Tumor Tissue. Thereafter, the labeled cell suspension was loaded onto an LS MACS column placed within a MidiMACS™ Separator (Miltenyi Biotec). The magnetically labeled CD133⁺ cells were retained in the column, while the unlabeled cells passed through, depleting the fraction of CD133⁺ cells. After removing the column from the magnetic field, the retained CD133⁺ cells were eluted as the positively selected fraction. To enhance purity, the positively selected fraction was subjected to a second round of separation using a new column.
Characterization of the isolated cells
-
Flow cytometric analysis.
To confirm that the isolated cells were CSCs, they were characterized using flow cytometric analysis for specific surface markers, including CD24, CD34, CD44, CD90, CD133, and CD326 (EpCAM). For staining, PE-conjugated CD24, CD44, CD90 and CD133 antibodies, as well as FITC-conjugated CD34 and CD326 antibodies were used. The cells were incubated with each antibody for 20 min in the dark at room temperature, following the manufacturer’s recommended concentrations. The stained cells were then analyzed using a Beckman Coulter Elite XL flow cytometer (California, USA) to confirm the expression of CSC surface markers, ensuring the successful isolation of hepatic CSCs12.
- Real time PCR analysis.
RNA was extracted from the isolated CSCs using the RNeasy Mini Kit (Cat#74104, Qiagen, Germany) following the manufacturer’s instructions. The concentration and purity of the extracted RNA were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Rockford, IL, USA), with the 260/280 nm absorbance ratio used to determine RNA quality. Complementary DNA (cDNA) synthesis was performed using the RevertAid First Strand cDNA Synthesis Kit (Cat# K1621, Thermo Fisher Scientific, Inc., Lithuania) according to the manufacturer’s protocol. For gene expression analysis, quantitative real-time PCR (qRT-PCR) was carried out using the DNA-Technology Real-Time PCR device (DT lite 4, Russia) to evaluate the expression levels of NOTCH1, NOTCH2, NOTCH3, and aldehyde dehydrogenase 1 A1 (ALDH1A1). The reaction mixture (25 µl volume) contained 12.5 µl of QuantiTect SYBR Green master mix (Cat# 204141, Qiagen, Germany), 0.75 µl of forward and reverse primer of target gene (Invitrogen, USA), 1.5 µl cDNA template and 9.5 µl RNase free water. β-actin was used as a housekeeping gene. Relative mRNA expression versus control value was assessed using the 2−ΔΔCt comparative method after normalization with β-actin gene.
The PCR cycling was set as follows: initial denaturation step at 95 °C for 15 min, followed by 40 cycles of denaturation at 94 °C for 15 s, annealing at 53 °C for NOTCH1 gene, 55 °C for NOTCH2 gene, 60 °C for NOTCH3 and ALDH1A1 genes for 30 s, and extension at 72 °C for 30 s. The primer sequences of each target gene are delineated in Table 1.
- Chemotherapy sensitivity assays.
The sensitivities of the isolated CSCs to chemotherapeutic drugs were evaluated utilizing the Cell Counting Kit-8 (CCK-8) assay (Sigma-Aldrich, USA), which is based on the WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2 H tetrazolium] colorimetric method. Briefly, 4000 cells/well were seeded in 96-well plates, and various concentrations (0.01, 0.1, 1, 10, and 100 µg/ml) of doxorubicin (Adricin, Doxorubicin Hcl, Hikma Specialized Pharmaceuticals, Egypt) were added at the beginning. After 72 h incubation, viable cells were measured by CCK-8 assay following the manufacturer’s instructions at 450 nm, using a microplate reader (Model 500; BIORed Instrument Inc., USA)48.
III. Evaluation of the anti-tumor efficacy of Q or K-loaded PLGA NPs with CD133 antibody against hepatic CSCs
Cytotoxic assay
The 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide is based on the conversion of MTT into formazan crystals by living cells, which reflects cytotoxicity based on mitochondrial activity49. The cytotoxic impact of the formulated NPs with CD133 antibody and loaded with the suggested natural compounds (Q or K) versus their free forms was measured by MTT assay using CSCs isolated from the Huh7 cell line. The cells were incubated with different concentrations of the compounds (20, 40, 80, and 160 µg/ml) for 24 h and 72 h at a cell density of 1 × 104 cells/well of a 96-well plate. After the different incubation time, MTT dissolved in PBS was added to each well at a final concentration of 5 mg/ml, and the samples were incubated at 37 °C for 4 h. After 4 hours, the medium was decanted and dimethyl sulfoxide (DMSO) was added to each well, including blank wells and left for 30 min to dissolve formazan crystals that formed during MTT cleavage in actively metabolizing cells. Absorbance of formazan in each plate was measured at 492 nm, using a microplate reader (Model 500; BIORad Instrument Inc., USA). For the untreated cells (negative control), medium was added instead of the test compound. All tests and analyses were done in triplicate, and the results were averaged.
Apoptotic assay
Annexin V and propidium iodide (PI) were applied for the determination of CSCs death mechanistic approach after treatments with the formulated NPs conjugated with CD133 antibody and loaded with Q or K and their free counterparts at 24 h of drug incubation. CSCs stained with only annexin V, only PI, or both dyes were considered as early apoptotic, necrotic, or late apoptotic cells, respectively. Therefore, the apoptotic analysis was dedicated to differentiating between early and late apoptotic cells, as well as necrotic cells. The apoptosis of the treated and untreated control cells was analyzed by flow cytometer (Beckman 230 Coulter Instrument, USA)50.
Metastatic (in vitro scratch) assay
In vitro scratch or wound-healing assay was used to assess the activity of the formulated NPs with CD133 antibody and loaded with Q or K and their free forms on the migration of hepatic CSCs. Cells were seeded in 12-well plates (2 × 105/well) with complete medium overnight to obtain a full confluent monolayer. After 24 h, the complete medium was removed and the confluent cell sheet was wounded through scratching the culture well surface with a 200 µl pipette tip. The scratch-wounded cells were washed twice with PBS to remove any cell fragments or detached cells before incubating in fresh medium with the tested compounds in either nanoforms or free forms for 24 h. Cell migration was monitored, and images of wound healing were captured by using the microscope after 24 h51.
Gene expression analysis
CSCs were seeded at a cell density of 4 × 104 cells/well and were treated with Q or K nanoformulations or their free forms for 48 h. Total RNA was isolated from cell samples using the RNAeasy mini Kit (Qiagen, Germany) then the concentration and purity of total extracted RNA were determined using a NanoDrop UV spectrophotometer (Thermo Fisher Scientific, USA). RNA of each treatment was converted to first-strand cDNA according to the manufacturer’s recommendations using Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Lithuania). Specific primer sequences (Table 2) for the drug resistance-related gene ATP-binding cassette superfamily G member 2 (ABCG2), P53, survivin, cyclin D1, c-Myc, vimentin, vascular endothelial growth factor (VEGF), and matrix metalloproteinase 7 (MMP-7) were used for quantitative real-time PCR. The reaction mixture (25 µl volume) included 12.5 µl of Maxima SYBR Green qPCR master mix (ThermoScientific, Lithuania), 0.75 µl of forward and reverse primers of the target gene (Invitrogen, USA), 1.5 µl cDNA template, and 9.5 µl RNase-free water. β-actin was used as a housekeeping gene. Relative mRNA expression versus control value was assessed using the 2−ΔΔCt comparative method after normalization with the β-actin gene.
The PCR cycling was set as follows: initial denaturation step at 95 °C for 15 min, followed by 40 cycles of denaturation at 94 °C for 15 s, annealing at 55 °C for 30 s, and extension at 72 °C for 30 s. DNA Technology Detecting Thermocycler DT Lite 4S1 (Russia) was used for gene expression quantitation.
Statistical analysis
The obtained results are represented by the means ± standard deviations (SD). Statistical analysis was performed using one-way analysis of variance (ANOVA) with the Statistical Package for the Social Sciences (SPSS), version 14. Post-hoc comparisons between groups were conducted using the least significant difference (LSD) test. A P-value < 0.05 was considered statistically significant.
Results
Characterization of the formulated NPs
Morphology and size of the prepared NPs using TEM
TEM images revealed that the synthesized NPs were spherical and conjugated with the CD133 antibody (Fig. 1). The average particle size of Q NPs with CD133 antibody ranged from 227 nm to 378 nm, whereas the K NPs with CD133 antibody exhibited an average size range of 202 nm to 321 nm.

Transmission electron microscope images of the prepared nanoparticles.
Particle size distribution and zeta potential of the prepared NPs
The particle size distribution and zeta potential of the synthesized NPs decorated with CD133 antibody were analyzed using a dynamic light scattering (DLS) instrument (Fig. 2). The Q NPs exhibited an average dynamic particle size of 234.8 nm and a negatively charged zeta potential of -14.5 mV. In comparison, the K NPs showed a larger average size of 336.4 nm and a zeta potential of -15.9 mV.

Particle size distribution and zeta potential charges of the prepared nanoparticles.
Fourier transform infrared spectroscopy (FTIR) analysis
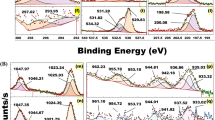
The chemical interactions between the ingredients in the loaded PLGA NPs with Q and K decorated with CD133 antibody were assessed using FTIR analysis. The functional groups of the components within the scanning range of 400 to 4000 cm− 1 exhibit substantial changes, as represented in Fig. 3. Alterations in the shape, position, and intensity of peaks were indicators of these interactions. PLGA polymer displays characteristic absorption bands at 1100–1250, and 1746–1760 cm-1, which represent the esters and carbonyl groups, respectively (Fig. 3a). Also, a hydroxyl group peak can be observed above 3000 cm-1. PLGA NPs spectrum depicts characteristic absorption bands at 1746 and 1230–1260 cm-1 which express the carbonyl groups and amide III, respectively (Fig. 3b). Either unloaded (Fig. 3c) or loaded (Fig. 3d and e) PLGA NPs decorated with CD133 antibody spectrum depicts characteristic absorption bands at 3450, 3276, 1746, 1630–1660, 1483–1540, and 1230–1260 cm-1 which represent the hydroxyl group, NH stretch, carbonyl group (lipid), carbonyl group (protein) with amide I, amide II, an d amide III, respectively.

FTIR spectra of PLGA [a], PLGA NPs [b], unloaded PLGA NPs decorated with targeted CD133 antibody [c], and loaded PLGA NPs with Q and K decorated with CD133 antibody [d and e].
Entrapment efficiency of Q and K within the NP
Entrapment efficiency of Q and K within the NPs decorated with CD133 antibody was assessed using dialysis tubing. The findings revealed that approximately 73% of Q and 83% of K were successfully encapsulated, based on their initial concentrations in the preparations (Table 3).
In vitro release profiles
The release profiles of the formulated Q and K NPs with CD133 antibody were assessed at pH 7.4 versus the dissolution behavior of the free drugs under identical conditions (Fig. 4). Free Q and K exhibited a rapid burst release, reaching nearly 100% within the first hour. In contrast, the nanoformulations of Q and K showed a more gradual release, with only 2% and 6% released after 1 h, increasing to 53% and 41%, respectively, over 24 h. These findings indicate that the NP system effectively reduces the initial burst release and offers a more controlled and sustained release profile in comparison with the free drug forms.
The drug release kinetic analysis revealed that the release profile of Q from the nanoparticle formulation was best described by the Higuchi model (R² = 0.990), indicating that the release mechanism is primarily diffusion-controlled. The Korsmeyer–Peppas model also showed a good fit, with an exponent n < 0.5, further supporting a Fickian diffusion mechanism. Meanwhile, the release profile of K from nanoparticles follows diffusion-controlled kinetics, as indicated by the best fit to the Higuchi model (R² = 0.988). The Korsmeyer–Peppas model also shows a good fit (R² = 0.976) with a diffusion exponent n < 0.5, suggesting a Fickian diffusion mechanism. In contrast, free Q and K showed immediate and complete release (~ 100% within 1 h), indicating no controlled release behavior.

Release profile of free Q and its nanoformula [A] as well as free K and its nanoformula [B] after dialysis against their solvent for 0, 1, 2, 3, 4, 5, and 24 h.
Characterization of the isolated cells
Flow cytometric analysis of the isolated cells from the Huh7 cell line
Flow cytometric analysis of the isolated cells from the Huh7 cell line revealed positive expression for CD24 (34.1%), CD34 (6.43%), CD44 (70.4%), CD90 (49.6%), CD133 (82.1%), and CD326 (64.4%) (Fig. 5).

Flow cytometric analysis of the isolated cells from the Huh7 cell line after staining with CD24, CD34, CD44, CD90, CD133, and CD326 antibodies.
Expression of CSC markers in the isolated cells from the Huh7 cell line
To further characterize the isolated CSCs, real-time PCR analysis was conducted to detect the expression levels of various CSC-related genes. The results demonstrated a significant (P < 0.05) upregulation of ALDH1A1, NOTCH1, and NOTCH3 mRNA expression in cells isolated from the Huh7 cell line compared to control cells (Fig. 6). In contrast, the expression of NOTCH2 showed an upregulation that was not statistically significant (P > 0.05) relative to the control (Fig. 6).

Expression of CSC-related genes in the isolated cells from Huh7 cell lines. All experiments were repeated 3 times. Data are the mean ± SD. * P < 0.05 compared with corresponding control cells.
Resistance of the isolated cells from Huh7 cell lines to chemotherapy
To assess the potential chemoresistance of the isolated cells from the Huh7 cell line, their sensitivity to different concentrations of doxorubicin was compared with that of control cells. The isolated cells exhibited markedly higher resistance to doxorubicin in comparison with control cells (Fig. 7).

Resistance of the isolated cells to doxorubicin. Data are the mean ± SD. * P < 0.05 compared with corresponding control cells.
Anti-cancer potential of Q and K NPs decorated with CD133 antibody
In vitro evaluation of the cytotoxic efficacy of the Q and K NPs decorated with CD133 antibody against hepatic CSCs
Figure 8 illustrates the cytotoxic effects of a broad concentration range (0–160 µg/ml) of both Q and K NPs and their free forms on CSCs isolated from the Huh7 cell line after 24 and 72 h of treatment. Notably, Q and its NP formulation exhibited significant (P < 0.05) cytotoxicity at concentrations of 40 µg/ml and 160 µg/ml, respectively, after 72 h, compared to control cells. In contrast, K and its NP form showed cytotoxic effects at 160 µg/ml and 20 µg/ml, respectively.

Cytotoxic impact of Q and K NPs and their free forms on CSCs isolated from Huh7 cell line after 24 and 72 h (results are mean ± SD of the triplicate experiments). * Significant change at P < 0.05 in comparison with control.
Apoptotic activity of Q and K NPs decorated with CD133 antibody against hepatic CSCs
Following the evaluation of the cytotoxic effect of free Q and free K, along with their nanoformulations (Q NPs and K NPs), against hepatic CSCs, this section explores the mechanistic approach of killing CSCs isolated from Huh7 cell lines. Flow cytometric analysis was employed for this purpose, as it is a widely used technique for distinguishing between necrosis, early apoptosis, and late apoptosis. This method allows for the assessment of individual cells within a population, providing more precise insights. As illustrated in Fig. 9, the percentage of cells in the Annexin V-negative/PI-negative quadrant (indicating viable cells) decreased to varying degrees following treatment with free Q and free K, and more prominently with their respective nanoformulations; Q NPs and K NPs.
Q NPs demonstrated the highest level of apoptosis induction, reaching 77.8%, compared to only 1.8% in the control—a highly significant increase. Detailed flow cytometric analysis revealed that Q NPs primarily induced late apoptosis (Annexin V-positive/PI-positive) in 43.17% of cells, followed by early apoptosis (Annexin V-positive/PI-negative) in 23.09%, along with an 11.58% increase in necrotic cells (Annexin V-negative/PI-positive). When compared to free Q, which induced apoptosis in only 12.01% of cells, Q NPs showed a markedly higher and statistically significant apoptotic effect. Interestingly, cell death induced by free Q occurred predominantly through early apoptosis, whereas Q NPs triggered a stronger late apoptotic response.
K and its nanoformulation induced apoptosis predominantly via early apoptotic pathways (Annexin V-positive/PI-negative). K NPs triggered an overall apoptotic response of 16.3%, which, significantly higher than the control (1.8%), while it was notably lower than that observed with Q NPs (77.8%). Specifically, K NPs induced 6.99% early apoptotic cells, 3.31% late apoptotic cells (Annexin V-positive/PI-positive), and 6.0% necrotic cells (Annexin V-negative/PI-positive). When compared to free K, which induced 24.64% total apoptosis, K NPs showed a slightly lower apoptotic effect that was not statistically significant. It is worth noting that free K primarily induced early apoptosis (14.85%), with only a small portion of the cell population progressing to late apoptosis—an essential step for effective CSCs eradication.

Apoptosis using flow cytometry of the proposed formulas. A] Schematic representation of quadrants for all treatments versus control. B] Graph of apoptotic versus unapoptotic cells upon treatments. C] Graph of necrotic, early apoptotic, and late apoptotic cells upon treatments.
The inhibitory effect of the Q and K NPs decorated with CD133 antibody on the migration of hepatic CSCs
The inhibitory effects of Q and K NPs and their free forms on the migration of CSCs isolated from the Huh7 cell line were assessed in vitro using the scratch wound-healing assay. As shown in Fig. 10, all treatments, including both free compounds and their nanoparticle formulations, effectively suppressed the migration of hepatic CSCs as indicated by the significant (P < 0.05) decrease in the wound closure area % after 24 h relative to control cells. Notably, the most pronounced inhibition was observed with Q NPs, indicating a strong anti-migratory potential.

Wound healing assay for demonstrating the inhibitory effect of the Q and K NPs and their free forms on the migration of hepatic CSCs at 24 h following wounding.
Gene expression levels of hepatic CSCs-related molecular pathways
Figure 11 illustrates the influence of Q and K NPs decorated with CD133 antibody and their free forms on the expression levels of several key genes in hepatic CSCs, including ABCG2, P53, survivin, vimentin, cyclin D1, c-Myc, MMP-7, and VEGF. The results confirmed that the treatment with Q NPs and K NPs led to a significant (P < 0.05) downregulation of ABCG2 gene expression level compared to the control cells. Additionally, all treatments exhibited a significant (P < 0.05) downregulation in the gene expression levels of survivin, vimentin, cyclin D1, MMP-7, and VEGF. Notably, all treatments—except free Q—significantly (P < 0.05) downregulated c-Myc gene expression level. In contrast, Q NPs and free K significantly (P < 0.05) upregulated P53 gene expression level, suggesting a potential role in promoting apoptosis and tumor suppression.

Influence of the Q and K NPs decorated with CD133 antibody and their free forms on the mRNA expression level of ABCG2, P53, survivin, vimentin, MMP-7, VEGF, cyclin D1, and c-Myc in Huh7 CSCs. Data are depicted as mean ± SD, Data were reproducible. *: Significant change at P < 0.05 compared with control cells.
Discussion
The theory of CSCs has marked a novel milestone in cancer research, as CSCs are believed to play a key role in tumor recurrence, metastasis, and resistance to chemotherapy59. Traditional stemness markers such as Nanog, SRY-box transcription factor 2 (SOX2), and Oct4 are commonly used to identify stem-like properties. In liver cancer, specific CSC markers include CD13, CD24, CD44, CD47, CD90, CD133, intercellular adhesion molecule 1 (ICAM1), EpCAM, and leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5)60. In the present study, cells isolated from the Huh7 cell line were found to express CD24 (34.1%), CD34 (6.43%), CD44 (70.4%), CD90 (49.6%), CD133 (82.1%), and CD326 (64.4%). These results fit with those of Chen et al.61, who suggested that cells co-expressing CD133 and EpCAM (CD326) are more indicative of CSCs within the Huh7 cell population. Moreover, hepatic CSCs have been reported to predominantly express CD44, which plays a crucial role in facilitating epithelial-mesenchymal transition (EMT)62. Furthermore, a previous study investigating the link between liver CSCs and early recurrence of HCC revealed that early recurrence was associated with the expression of CD9063. Meanwhile, it has been documented that CD24⁺ hepatoma cells also express CD133 and CD326, with CD24 expression overlapping with these markers. This overlap suggests that CD24, CD133, and CD326 may share similar self-renewal characteristics, highlighting their potential role in identifying cancer stem-like cells64.
The current study revealed that hepatic CSCs isolated from the Huh7 cell line showed significant upregulation in the transcriptional levels of NOTCH1, NOTCH3, and ALDH1A, while NOTCH2 exhibited a non-significant increase. The Notch signaling pathway, known for being evolutionarily conserved, plays a critical role in regulating CSC proliferation, self-renewal, differentiation, angiogenesis, and migration65. More importantly, the Notch signaling pathway has been reported to be a key player in the acquisition of EMT, which is critically linked to drug resistance. This suggests that targeting the Notch pathway could offer a therapeutic approach for treating cancer by overcoming drug resistance in cancer cells. Such an approach may help eliminate CSCs or EMT-like cells, which are believed to be responsible for tumor relapse66. It has been found that NOTCH2 and JAGGED1 are significantly upregulated in gemcitabine-resistant pancreatic cancer cells. Notch signaling downregulation resulted in a partial reversal of the EMT phenotype, inducing a mesenchymal-to-epithelial transition (MET). This transition was associated with a decrease in the expression of vimentin, zinc finger E-box-binding homeobox 1 (ZEB1), Slug, Snail, and NF-κB expression, all of which are key regulators of the EMT process67. Persistent activation of the Notch signaling pathway has been suggested to play a crucial role in both reprogramming hepatic progenitor cells (HPCs) into CSCs and in maintaining CSC characteristics throughout HCC development68. Luo et al.69 indicated that the Notch pathway is turned on in CD90⁺ liver CSCs, suggesting that the Notch pathway may participate in HCC carcinogenesis. Additionally, it has been reported that ABCG2-positive glioma stem cells exhibit high chemoresistance and preferentially express the NOTCH1 gene70. In HCC models, ablation of NOTCH3 has been shown to exacerbate the apoptotic response to doxorubicin, a process that is largely dependent on p5371. High expression of NOTCH2 has been observed in HCC cells that express CD9072. Functional studies have revealed that knockdown of NOTCH2 leads to reduced cell proliferation, impaired cell cycle progression, and decreased colony formation in HepG2 cells. These findings suggest that NOTCH2 is essential for the proliferation and self-renewal of HCC cells73.
Earlier studies have shown that cancer cells with high ALDH activity exhibit enhanced tumorigenic capacity and chemoresistance across different cancer types. Specifically, a subpopulation of cells with high ALDH activity has been identified in highly tumorigenic colon CSCs, which display a stem-like EpCAM⁺/CD44⁺ phenotype74. Moreover, in breast CSCs, the high ALDH activity has been correlated with more aggressive tumor behavior and increased chemoresistance. As a result, it may serve as a marker for poor clinical outcomes75. Ma et al.76 have observed that ALDH-positive cells highly express the primitive cell surface marker CD133, suggesting that ALDH could serve as a positive marker for tumorigenic HCC CSCs. These investigators found that CD133⁺ALDH⁺ cells are more tumorigenic than CD133⁺ALDH⁻ cells when grafted into mice. Given these findings, it is possible to propose that ALDH may act as a novel marker of poor prognosis and a potential target for HCC therapy. Additionally, silencing Snail expression significantly downregulates ALDH1 expression, thereby suppressing the stem-like characteristics and in vivo tumorigenic activity of CD44⁺CD24⁻ALDH1⁺ cells77.
Following the molecular genetic findings, the isolated cells from the Huh7 cell line in the current study showed a much higher resistance to doxorubicin. This observation aligns with the results of Ma et al.78. Growing evidence indicates that the CSC population plays a key role in chemoresistance and cancer relapse, as it has the ability to self-renew and differentiate into various cancer cell lineages in response to chemotherapy79. CSCs are also able to trigger cell cycle arrest (a quiescent state), which contributes to their resistance to chemotherapy and radiotherapy80. ATP-binding cassette (ABC) transporters are able to release a wide variety of toxin-producing materials from cells, thereby reducing the effectiveness of drugs in killing cancer cells and directly contributing to the development of drug resistance. It has been reported that CSCs overexpress ABC transporters and dysregulate signaling pathway networks, which together contribute to the acquisition of multidrug resistance and the maintenance of self-renewal characteristics81.
The dysregulation of Notch, TGF-β, and Wnt/β-catenin signaling pathways has been reported to play a key role in the evolution of CSCs, contributing to the acquisition and maintenance of stem-like traits such as self-renewal, plasticity, and quiescence60. Cancer stem cells can be effectively eliminated by targeting core signaling pathways. Given the crosstalk between CSCs and HCC cells, complex CSC niches play a crucial role in tumor growth and resistance to treatment. Single or combination therapies that target these CSC niches have shown effectiveness in treating CSCs. Specifically, targeting CSC surface markers offers a direct approach to addressing these cells82.
In the current study, Q and K PLGA NPs functionalized with CD133 antibody were developed to specifically target hepatic CSCs expressing CD133 antigen. The average particle size of the Q NPs with CD133 antibody was approximately 227 nm, with a negative zeta potential of -14.5 mV. The K NPs with CD133 antibody had an average particle size of around 336.4 nm and a negative zeta potential of -15.9 mV. These findings are consistent with a previous study that reported salinomycin CD133-NPs with a suitable size of 149.2 nm and sustained drug release, exhibiting a negative zeta potential of -22.8 mV, similar to our Q NPs with CD133 antibody83.
The chemical interactions between the components in the loaded PLGA NPs with Q or K, decorated with the targeted CD133 antibody, were investigated using FTIR scanning. The alterations in the shape, position, and intensity of the peaks served as indicators of these interactions84. The PLGA polymer exhibits characteristic absorption bands in the range of 1100–1250 cm⁻¹, corresponding to ester groups, and at 1746–1760 cm⁻¹, representing carbonyl (C = O) groups. Additionally, a broad peak indicating the presence of hydroxyl (–OH) groups can be observed above 3000 cm⁻¹85,86. Furthermore, the FTIR spectrum of PLGA NPs shows characteristic absorption bands at 1746 cm⁻¹ and 1230–1260 cm⁻¹, corresponding to carbonyl groups and Amide III, respectively. In both unloaded and drug-loaded PLGA NPs decorated with the targeted CD133 antibody, the spectrum displays distinct absorption bands at 3450 cm⁻¹ (hydroxyl group), 3276 cm⁻¹ (N–H stretch), 1746 cm⁻¹ (carbonyl group—lipid), 1630–1660 cm⁻¹ (carbonyl group—protein/Amide I), 1483–1540 cm⁻¹ (Amide II), and 1230–1260 cm⁻¹ (Amide III)86,87. These bands confirm the presence of specific functional groups and successful decoration with the antibody.
The entrapment efficiency of the Q NPs and K NPs with CD133 antibody coupled to their surfaces was approximately 73% and 83%, respectively. Notably, the entrapment efficiency of our formulations was higher than that reported for salinomycin CD133-NPs, which had an efficiency of 63.2%83. The nanoformulations of Q and K showed a more gradual release, with only 2% and 6% released after 1 h, increasing to 53% and 41%, respectively, over 24 h. These findings indicate that the NP system effectively reduces the initial burst release and offers a more controlled and sustained release profile in comparison with the free drug forms. The burst release could be attributed to the drug adsorbed onto or close to the surface of the nanoparticles, and also due to the high surface-to-volume ratio of the nanoparticle88.
The formulated NPs were further assessed for their potential to induce cytotoxicity, promote apoptosis, inhibit metastasis, reverse drug resistance, and interfere with CSC-associated signaling pathways in Huh7 CSCs, in comparison to their free forms. The results revealed that both Q and Q-loaded NPs with CD133 antibody exerted significant cytotoxic effects on Huh7 CSCs at concentrations of 40 µg/ml and 160 µg/ml, respectively, after 72 h. Notably, Q NPs with CD133 antibody showed the most pronounced apoptotic effect, inducing apoptosis in 77.8% of cells. Specifically, Q NPs with CD133 antibody primarily triggered late apoptosis (43.17%), followed by early apoptosis (23.09%), along with an 11.58% increase in necrotic cells. In contrast, free Q induced cell death predominantly through early apoptosis rather than late apoptosis, as seen with Q NPs with CD133 antibody. Hisaka et al.89 recorded that quercetin has been shown to suppress liver cancer cell proliferation via promoting apoptosis and causing cell cycle arrest. Studies have reported that Q promotes apoptosis through the upregulation of the tumor suppressor protein p53 and the downregulation of cyclin D1. Additionally, it has been observed to induce cell cycle arrest at the S phase, further contributing to its antiproliferative effects90. Q also exerts its antiproliferative and pro-apoptotic effects via promoting cell cycle arrest and apoptosis through the phosphorylation of ERK and JNK within the MAPK signaling pathway. Furthermore, it disrupts the PI3K/AKT pathway by promoting the phosphorylation of PI3K, AKT, and S6K, enhancing its anti-cancer activity. Q modulates the Wnt/β-catenin pathway by prohibiting β-catenin nuclear translocation and inhibiting the production of MMPs, which are essential for tumor cell migration. Additionally, it suppresses the JAK/STAT pathway by blocking the formation of phosphorylated STAT (p-STAT), reinforcing its antiproliferative properties. Notably, treatment with Q NPs decorated with CD133 antibody has been shown to effectively inhibit liver cancer cell proliferation, migration, and colony formation, thereby significantly repressing the progression of liver cancer91.
The cytotoxic effects of K and its PLGA nanoformulation with CD133 antibody against Huh7 CSCs were evident at concentrations of 160 µg/ml for free K and 20 µg/ml for K NPs with CD133 antibody. Both K and K NPs with CD133 antibody primarily induced apoptosis through early apoptotic mechanisms. Specifically, K NPs with CD133 antibody led to 6.99% early apoptotic cells, followed by 3.31% late apoptosis, and a 6.0% increase in necrotic cells. Overall, K NPs with CD133 antibody induced apoptosis in 16.3% of cells, which was slightly lower and not statistically significant compared to the 24.64% observed with free K. Notably, free K induced cell death mainly through early apoptosis (14.85%), with only a small portion of the cell population advancing to late apoptosis—an important phase for ensuring complete elimination of CSCs. Mylonis et al.92 registered that K treatment of Huh7 cells resulted in a marked reduction in cell viability, with this effect being significantly more pronounced under hypoxic conditions. Additionally, K was found to induce G2/M phase cell cycle arrest, accompanied by a decrease in the expression levels of cyclin B and CDK1 in treated cancer cells. Furthermore, K treatment led to a concentration-dependent increase in acidic vesicular organelle (AVO)-positive cells compared to the control group. These findings suggest that K not only impairs cell proliferation but also induces autophagy in Huh7 liver cancer cells93. K also contributes to its anticancer activity by activating tumor suppressor genes and inhibiting key pathways involved in cancer progression. These include the suppression of angiogenesis, the PI3K/AKT signaling pathway, STAT3, the transcription factor AP-1, Nrf2, and other critical cell signaling molecules. Through this multifaceted interference, K disrupts cancer cell survival, proliferation, and metastasis94.
The wound healing assay results of this study demonstrated that the migration of hepatic CSCs was effectively inhibited by both the proposed NPs with CD133 antibody and their free drug counterparts, with the most significant anti-migratory effect observed following treatment with Q NPs with CD133 antibody. Previous studies have reported that Q promotes the proteasomal degradation of Ras homolog family member C (RhoC), thereby suppressing cell migration and invasion in Huh7 cells. This mechanism likely contributes to the pronounced inhibition of CSCs migration observed in this work95. Furthermore, the data from Hung et al.96 study suggested that K can suppress the invasion and migration of RCC786-O cells in vitro by downregulating the expression of MMP-2. This effect is achieved through the inhibition of focal adhesion kinase (FAK) and Akt phosphorylation, highlighting K potential to interfere with key signaling pathways involved in cancer cell motility and metastasis. Also, Ju et al.97 found that K treatment (25 µM) resulted in a significant reduction in cell migration in both Huh-7 and SK-Hep-1 cells, compared to the vehicle control. This inhibitory effect on migration was attributed to the down-regulation of MMP-9 expression and the inhibition of AKT signaling, which are both crucial for the cell’s migratory and invasive capabilities.
In this study, both Q NPs and K NPs with CD133 antibody led to a marked downregulation of ABCG2 gene expression in Huh7 CSCs. This result is consistent with earlier reports suggesting that phytochemical-based nanoformulations can effectively suppress ABC transporter expression, particularly ABCG2, which is often associated with drug resistance and CSCs maintenance98,99. Such downregulation may contribute to overcoming chemoresistance and improving therapeutic outcomes100. Breast cancer resistance protein (BCRP, also known as ABCG2) belongs to the ATP-binding cassette G family and plays a significant role in the efflux of various anticancer drugs, thereby contributing to the development of chemoresistance, particularly in digestive system cancers. ABCG2 expression is notably elevated in the side population (SP) of stem-like cells, which exhibit enhanced drug resistance compared to non-stem-like cancer cells, highlighting its importance in maintaining the chemoresistant phenotype of CSCs101. Interestingly, ABCG2 expression has been closely associated with key aspects of HCC progression, including tumor initiation, cell proliferation, metastasis, and the development of chemoresistance102. Thus, BCRP (ABCG2) serves as a potential marker for liver CSCs103,104. Namisaki et al.105 recorded that ABCG2 expression, which is subject to modulation by the AKT signaling pathway, has a substantial role in mediating the efflux of chemotherapeutic agents like doxorubicin from HCC cells, thereby reducing their therapeutic effectiveness. Moreover, the emergence of a liver CSCs phenotype—characterized by elevated ABCG2 expression—is strongly linked to malignant behaviors, including enhanced proliferation, migration, and invasion, further highlighting ABCG2’s involvement in HCC aggressiveness and treatment resistance104. Notably, these malignant characteristics can be significantly diminished by downregulating ABCG2 expression. This highlights the potential therapeutic value of targeting ABCG2 to impair liver CSC traits and enhance the effectiveness of anticancer treatments in HCC103.
Epithelial-mesenchymal transition is recognized as a key mechanism driving chemoresistance in HCC. During this process, HCC cells exhibit downregulation of epithelial markers such as desmoplakin, E-cadherin, and claudin-1, which are essential for maintaining cell-cell adhesion and epithelial integrity. Concurrently, there is an upregulation of mesenchymal markers and EMT-related factors, including neural (N)-cadherin, vimentin, matrix metalloproteinases, and several transcription factors such as Snail1, Snail2, ZEB1, ZEB2, and Twist. This phenotypic shift enhances cell motility, invasiveness, and contributes to drug resistance, ultimately facilitating tumor progression and metastasis106. Furthermore, EMT initiates the activation of several key signaling pathways that are pivotal in promoting tumor progression and therapy resistance. These include the transforming growth factor (TGF)-β/SMAD pathway, Wnt/β-catenin signaling, the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) cascade, the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) axis, and the Notch signaling pathway107. These EMT-induced alterations markedly enhance the resistance of HCC cells to apoptosis and chemotherapeutic agents. A critical link between angiogenesis and HCC progression lies in the epithelial-to-mesenchymal transition of liver epithelial cells, which is often driven by angiogenic factors—particularly VEGF. VEGF not only promotes neovascularization but also facilitates EMT, thereby accelerating tumor growth, invasion, and resistance to treatment108. Furthermore, vimentin filaments play a crucial role in protecting cancer cells from mechanical stresses encountered during migration or when squeezing through confined spaces. By supplying a viscoelastic framework, vimentin ensures the structural integrity of the cell, particularly the positioning and stability of organelles, including the nucleus. This function is essential during EMT and cancer progression, as it helps cancer cells maintain their shape and adapt to the physical challenges of invasion and metastasis109.
MMP-7 has been extensively studied in cancer progression due to its dual role in both extracellular matrix degradation and metastasis promotion, as well as its involvement in the regulation of the Fas/FasL system and apoptosis sensitivity in tumor cells. MMP-7 influences Fas expression and activation by cleaving the membrane-bound form of FasL, generating its soluble form110, and by cleaving the Fas receptor itself. In both instances, MMP-7 activity effectively blocks the induction of apoptosis by Fas activation. This protective mechanism allows tumor cells to evade apoptosis, contributing to enhanced cell survival, resistance to chemotherapy, and overall tumor progression111. Furthermore, survivin has been identified as a downstream gene of the Wnt/β-catenin signaling pathway, which has a fundamental role in the regulation of gastric CSCs. The activation of this pathway promotes the expression of survivin, a protein that inhibits apoptosis and supports cell survival, contributing to the maintenance of CSC properties such as self-renewal, tumorigenicity, and resistance to chemotherapy112. It has been recorded that glioma stem cells (GSCs) contribute to treatment resistance in tumor cells by upregulating DNA damage checkpoint proteins113. CSCs and survivin are widely recognized as key factors contributing to the recurrence of tumors, and radiation and drug resistance observed in recurrent tumors114. On the other hand, TGF-β/Smad signaling plays a critical role as an effector in various signaling pathways regulating the self-renewal of both normal and CSCs. Cyclin D1 serves as a key regulator of early cell fate decisions during the G1 phase of human embryonic stem cells (hESCs) by controlling the transcriptional activity of Smad2/3115.
In the current study, the treatment of Huh7 CSCs with Q, Q NPs with CD133 antibody, K, and K NPs with CD133 antibody downregulated the expression levels of VEGF, vimentin, MMP-7, survivin, and cyclin D1 genes. These results are in agreement with those of previous studies, which demonstrated the anti-angiogenic, anti-metastatic, and pro-apoptotic effects of Q116 and K117,118 in various cancer models. For instance, it has been reported that Q suppresses VEGF expression and angiogenesis by modulating HIF-1α and PI3K/Akt signaling119. Furthermore, Wu et al.120 clarified that Q has been reported to reverse invasion and migration by modulating EMT-related markers. Specifically, it upregulated E-cadherin—a key epithelial marker—while downregulating mesenchymal markers like N-cadherin and vimentin at both gene and protein expression levels in HCC LM3 cells. This shift from a mesenchymal to an epithelial phenotype suggests that Q can suppress EMT, thereby limiting cancer cell migration, invasiveness, and possibly reducing the stem-like properties associated with cancer progression and resistance. While, Lee et al.121 stated that, Q dose-dependently inhibited the transcriptional activity of specificity protein 1 (Sp1) and subsequently downregulated the expression of its downstream targets, including p27, p21, cyclin D1, Mcl-1, and survivin in HepG2 cells. This suggests that Q can interfere with Sp1-mediated transcriptional regulation, leading to cell cycle arrest and apoptosis. In addition, Q NPs were found to upregulate p27, a cyclin-dependent kinase inhibitor that plays a crucial role in inducing cell cycle arrest in liver cancer cells. Moreover, treatment with Q NPs led to the significant, dose-dependent inhibition of several key oncogenic and cell cycle-related proteins, including c-Myc, cyclin-D1, CDK1, MMP-7, and β-catenin32. These molecular changes contributed to the induction of apoptosis in liver cancer cells, further supporting the therapeutic potential of Q nanoformulations in targeting liver CSCs and overcoming chemoresistance.
It was shown that K inhibits the expression of VEGF in ovarian cancer cells, thereby reducing angiogenesis, which is crucial for tumor growth and metastasis122. Zhu et al.123 found that K dramatically suppressed the viability, proliferation, migration, and invasion of HepG2 liver cancer cells. Notably, K treatment led to a significant downregulation in the expression levels of cyclin D1, MMP-2, MMP-9, and vimentin124,125. Colombo et al.126 demonstrated that, K effectively suppressed glioma cell growth and migration when administered via K-loaded nanoemulsion and mucoadhesive nanoemulsion formulations.
c-Myc is indeed a critical “master” transcription factor that orchestrates the expression of a broad spectrum of genes essential for various cellular processes, including cell proliferation, differentiation, angiogenesis, apoptosis, metabolism, EMT, invasion, and metastasis127. Absolutely, c-Myc has been shown to regulate the expression of vimentin and associated pathways for the induction of EMT and cell migration128. Previous research has demonstrated that the deletion of the tumor suppressor gene p53 promotes hepatocyte proliferation and tumor formation129. In addition to the deletion of p53, the Bcl-2 and Bmi-1 overexpression, along with p19ARF loss, further aid Myc in regulating the survival and proliferation of CSCs130,131.
In the current investigation, Q NPs with CD133 antibody, K, and K NPs with CD133 antibody were found to significantly downregulate c-Myc expression levels in Huh7 CSCs. Moreover, treatment of Huh7 CSCs with Q NPs with CD133 antibody and K led to a significant upregulation of P53 gene expression. These findings are consistent with the results reported by previous studies, which demonstrated that Q and K can modulate critical oncogenes and tumor suppressor genes involved in cancer progression and stemness maintenance132,133. Shahbaz et al.133 declared that Q exerted multiple antitumor effects on pancreatic cancer cells, including the apoptosis induction, proliferation inhibition, migration and invasion suppression, and metastasis and tumor growth reduction in pancreatic ductal adenocarcinoma (PDA) xenograft models. These effects are largely attributed to its ability to downregulate c-Myc expression, which significantly limits the proliferative capacity of pancreatic cancer cells, while simultaneously upregulating p53. Furthermore, Zhang and Ma132 mentioned that K enhanced the efficacy of cisplatin by modulating key regulatory genes involved in cell cycle control and apoptosis. Specifically, K reduces c-Myc mRNA levels while upregulating cyclin-dependent kinase inhibitor 1 A (CDKN1A), which plays a crucial role in arresting the cell cycle. Interestingly, in breast cancer MDA-MB-453 cells, K appears to enhance p53 expression, suggesting a dual mechanism of action—both suppressing oncogenic signals and promoting tumor suppressor pathways.
In conclusion, the findings of this study spotlight the effective targeting of hepatic CSCs by CD133 antibody surface-modified quercetin and kaempferol NPs. This efficacy was achieved through their capacity to induce apoptosis, inhibit metastasis, overcome drug resistance, and modulate key CSCs-related signaling pathways. Hence, nanoparticle surface modification with CD133 antibody emerges as a highly beneficial strategy for enhancing therapeutic properties of the anti-cancer agents, rendering these NPs multifunctional and particularly well-suited for targeting CSCs in hepatocellular carcinoma.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Kinsey, E. & Lee, H. M. Management of hepatocellular carcinoma in 2024: the multidisciplinary paradigm in an evolving treatment landscape. Cancers 16 (3), 666 (2024).
Sathishbabu, P. et al. Current advances in nanoparticle-based approaches for the hepatocellular carcinoma treatment. Clinics Res. Hepatol. Gastroenterol. 102508 (2024).
Ali, A. A. K. et al. Assessment of clinico-epidemiological profile of hepatocellular carcinoma in the last two decades. Egypt. J. Intern. Med. 35 (1), 18 (2023).
Omar, A. et al. Egyptian society of liver cancer recommendation guidelines for the management of hepatocellular carcinoma. J. Hepatocell. Carcinoma 1547–1571 (2023).
Okeke, E. et al. Epidemiology of liver cancer In Africa: Current and future trends. In Seminars In Liver Disease (Thieme Medical, 2020).
Albarrak, J. & Al–Shamsi, H. Current status of management of hepatocellular carcinoma in the Gulf region: Challenges and recommendations. Cancers 15 (7) (2001).
Phi, L. T. H. et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018(1), 5416923 (2018).
Lee, T. K. W., Guan, X. Y. & Ma, S. Cancer stem cells in hepatocellular carcinoma—from origin to clinical implications. Nat. Reviews Gastroenterol. Hepatol. 19 (1), 26–44 (2022).
Taib, N. et al. Treatment with decitabine induces the expression of stemness markers, PD-L1 and NY-ESO-1 in colorectal cancer: potential for combined chemoimmunotherapy. J. Translational Med. 21 (1), 235 (2023).
Jang, J. W. et al. CD133 confers cancer stem-like cell properties by stabilizing EGFR-AKT signaling in hepatocellular carcinoma. Cancer Lett. 389, 1–10 (2017).
Xi, G. et al. Targeting CD133 improves chemotherapeutic efficacy of recurrent pediatric pilocytic Astrocytoma following prolonged chemotherapy. Mol. Cancer. 16, 1–11 (2017).
Ma, S. et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 132 (7), 2542–2556 (2007).
Zhang, L. et al. Inhibitory effects of transcription factor Ikaros on the expression of liver cancer stem cell marker CD133 in hepatocellular carcinoma. Oncotarget 5 (21), 10621 (2014).
Chen, K., Huang, Y. & Chen, J. -l. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol. Sin. 34 (6), 732–740 (2013).
Sun, Y. et al. Targeting cancer stem cells with polymer nanoparticles for Gastrointestinal cancer treatment. Stem Cell Res. Ther. 13 (1), 489 (2022).
Fernandes, Q. et al. Shrinking the battlefield in cancer therapy: Nanotechnology against cancer stem cells. Eur. J. Pharm. Sci. 106586 (2023).
Lyakhovich, A. & Lleonart, M. E. Bypassing mechanisms of mitochondria-mediated cancer stem cells resistance to chemo‐and radiotherapy. Oxid. Med. Cell. Longev. 1, 1716341 (2016).
Zhang, Z. et al. Nanoparticles and the treatment of hepatocellular carcinoma. Nanatechnol. Reviews. 13 (1), 20240134 (2024).
Fu, Z. & Xiang, J. Aptamer-functionalized nanoparticles in targeted delivery and cancer therapy. Int. J. Mol. Sci. 21 (23), 9123 (2020).
Li, F. et al. Enhanced cellular internalization and on-demand intracellular release of doxorubicin by Stepwise pH-/reduction-responsive nanoparticles. ACS Appl. Mater. Interfaces. 8 (47), 32146–32158 (2016).
Shi, Y., van der Meel, R., Chen, X. & Lammers, T. The EPR effect and beyond: strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 10 (17), 7921 (2020).
Guo, Y. et al. Mechanisms of chemotherapeutic resistance and the application of targeted nanoparticles for enhanced chemotherapy in colorectal cancer. J. Nanobiotechnol. 20 (1), 371 (2022).
Kong, F. H. et al. Current status of Sorafenib nanoparticle delivery systems in the treatment of hepatocellular carcinoma. Theranostics 11 (11), 5464 (2021).
Yoo, J. et al. Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers 11 (5), 640 (2019).
Boateng, F. & Ngwa, W. Delivery of nanoparticle-based radiosensitizers for radiotherapy applications. Int. J. Mol. Sci. 21 (1), 273 (2019).
Li, B., Li, Q., Mo, J. & Dai, H. Drug-loaded polymeric nanoparticles for cancer stem cell targeting. Front. Pharmacol. 8, 51 (2017).
Tian, H. et al. Enhancing the therapeutic efficacy of nanoparticles for cancer treatment using versatile targeted strategies. J. Hematol. Oncol. 15 (1), 132 (2022).
Poonaki, E., Nickel, A. C., Ardestani, S. & Rademacher, M. CD133-functionalized gold nanoparticles as a carrier platform for Telaglenastat (CB-839) against tumor stem cells. Int. J. Mol. Sci. 23 (10), 5479 (2022).
Taylor, W. F. & Jabbarzadeh, E. The use of natural products to target cancer stem cells. Am. J. Cancer Res. 7 (7), 1588 (2017).
Michala, A. S. & Pritsa, A. Quercetin: a molecule of great biochemical and clinical value and its beneficial effect on diabetes and cancer. Diseases 10 (3), 37 (2022).
Batsukh, T. & Tsend-Ayush, A. Herbal drug–based nanotherapy for hepatocellular carcinoma: Quercetin–contained nanocarrier as a multipurpose therapeutic agent against hepatocellular carcinoma. Biomedical Rep. 22 (2), 29 (2024).
Ren, K. W. et al. Quercetin nanoparticles display antitumor activity via proliferation Inhibition and apoptosis induction in liver cancer cells. Int. J. Oncol. 50 (4), 1299–1311 (2017).
Singh, T. et al. The role of phytonutrient Kaempferol in the prevention of Gastrointestinal cancers: recent trends and future perspectives. Cancers 16 (9), 1711 (2024).
Alam, W. et al. Kaempferol as a dietary anti-inflammatory agent: current therapeutic standing. Molecules 25 (18), 4073 (2020).
Ashrafizadeh, M. et al. Therapeutic effects of Kaempferol affecting autophagy and Endoplasmic reticulum stress. Phytother. Res. 34 (5), 911–923 (2020).
Deng, S. P. et al. Synthesis, spectroscopic study and radical scavenging activity of Kaempferol derivatives: enhanced water solubility and antioxidant activity. Int. J. Mol. Sci. 20 (4), 975 (2019).
Dara, T. et al. Erythropoietin-loaded solid lipid nanoparticles: preparation, optimization, and in vivo evaluation. Colloids Surf., B. 178, 307–316 (2019).
Liang, M., Li, L. D., Li, L. & Li, S. Nanotechnology in diagnosis and therapy of Gastrointestinal cancer. World J. Clin. Cases. 10 (16), 5146 (2022).
Alyami, N. M., Alyami, H. M. & Almeer, R. Using green biosynthesized kaempferol-coated sliver nanoparticles to inhibit cancer cells growth: an in vitro study using hepatocellular carcinoma (HepG2). Cancer Nanotechnol. 13 (1), 26 (2022).
Mohd-Zahid, M. H. et al. Gold nanoparticles conjugated with anti-CD133 monoclonal antibody and 5-fluorouracil chemotherapeutic agent as nanocarriers for cancer cell targeting. RSC Adv. 11 (26), 16131–16141 (2021).
Kuldeep, G. Formulation and charcterization of Kaempferol nanoparticles. Asian J. Biomedical Pharm. Sci. 4, 59–63 (2014).
Anwer, M. K. et al. Development and evaluation of PLGA polymer based nanoparticles of Quercetin. Int. J. Biol. Macromol. 92, 213–219 (2016).
Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 52 (12), 1145–1149 (1963).
Korsmeyer, R. W. et al. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 15 (1), 25–35 (1983).
Xiao, W. et al. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J. Experimental Clin. Cancer Res. 36, 1–13 (2017).
Gangavarapu, K. J., Miller, A. & Huss, W. J. Gene expression in single cells isolated from the CWR-R1 prostate cancer cell line and human prostate tissue based on the side population phenotype. Single Cell Biol. 5(3) (2016).
Song, Y. et al. Altered expression of PTCH and HHIP in gastric cancer through their gene promoter methylation: novel targets for gastric cancer. Mol. Med. Rep. 7 (4), 1159–1168 (2013).
Li, J. et al. Establishment of a novel system for the culture and expansion of hepatic stem-like cancer cells. Cancer Lett. 360 (2), 177–186 (2015).
Van Meerloo, J., Kaspers, G. J. L. & Cloos, J. Cell sensitivity assays: The MTT assay. Cancer Cell. Culture: Methods Protoc. 237–245 (2011).
Liu, Y. et al. A novel matrine derivate inhibits differentiated human hepatoma cells and hepatic cancer stem-like cells by suppressing PI3K/AKT signaling pathways. Acta Pharmacol. Sin. 38 (1), 120–132 (2017).
Liang, C. C., Park, A. Y. & Guan, J. L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2 (2), 329–333 (2007).
Sukowati, C. H. C. et al. Gene and functional up-regulation of the BCRP/ABCG2 transporter in hepatocellular carcinoma. BMC Gastroenterol. 12, 1–8 (2012).
Fan, Y. et al. miR-19b promotes tumor growth and metastasis via targeting TP53. RNA 20 (6), 765–772 (2014).
Yu, J. et al. Increased expression of survivin in gastric cancer patients and in first degree relatives. Br. J. Cancer. 87 (1), 91–97 (2002).
Liu, J. et al. Enhancement of canonical Wnt/β-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. PLoS One. 6 (11), e27496 (2011).
Vedagiri, D. et al. An atypical system for studying epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 6 (1), 26282 (2016).
Liu, L. Z. et al. Role and mechanism of arsenic in regulating angiogenesis. PLoS One 6(6), e20858 (2011).
Ozaki, I. et al. Involvement of the Ets-1 gene in overexpression of matrilysin in human hepatocellular carcinoma. Cancer Res. 60 (22), 6519–6525 (2000).
Xia, P. & Liu, D. H. Cancer stem cell markers for liver cancer and pancreatic cancer. Stem Cell. Res. 60, 102701 (2022).
Zhou, H. M., Zhang, J. G., Zhang, X. & Li, Q. Targeting cancer stem cells for reversing therapy resistance: mechanism, signaling, and prospective agents. Signal. Transduct. Target. Therapy. 6 (1), 62 (2021).
Chen, Y. et al. CD133(+)EpCAM(+) phenotype possesses more characteristics of tumor initiating cells in hepatocellular carcinoma Huh7 cells. Int. J. Biol. Sci. 8 (7), 992–1004 (2012).
Okabe, H. et al. CD44s signals the acquisition of the mesenchymal phenotype required for anchorage-independent cell survival in hepatocellular carcinoma. Br. J. Cancer. 110 (4), 958–966 (2014).
Guo, Z. et al. Cancer stem cell markers correlate with early recurrence and survival in hepatocellular carcinoma. World J. Gastroenterology: WJG. 20 (8), 2098 (2014).
Ji, J. & Wang, X. W. Clinical implications of cancer stem cell biology in hepatocellular carcinoma. In Seminars in Oncology. (Elsevier, 2012).
Wang, X. et al. Musashi2 contributes to the maintenance of CD44v6 + liver cancer stem cells via notch1 signaling pathway. J. Experimental Clin. Cancer Res. 38 (1), 1–19 (2019).
Wang, Z. et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim. Et Biophys. Acta (BBA)-Reviews Cancer. 1806 (2), 258–267 (2010).
Wang, Z. et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the Notch signaling pathway. Cancer Res. 69 (6), 2400–2407 (2009).
Geisler, F. & Strazzabosco, M. Emerging roles of Notch signaling in liver disease. Hepatology 61 (1), 382–392 (2015).
Luo, J. et al. The Notch pathway promotes the cancer stem cell characteristics of CD90 + cells in hepatocellular carcinoma. Oncotarget 7 (8), 9525 (2015).
Patrawala, L. et al. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2 + and ABCG2 – cancer cells are similarly tumorigenic. Cancer Res. 65 (14), 6207–6219 (2005).
Giovannini, C., Bolondi, L. & Gramantieri, L. Targeting Notch3 in hepatocellular carcinoma: molecular mechanisms and therapeutic perspectives. Int. J. Mol. Sci. 18 (1), 56 (2016).
Sukowati, C. H. C. et al. The expression of CD90/Thy-1 in hepatocellular carcinoma: an in vivo and in vitro study. PLoS One. 8 (10), e76830 (2013).
Wu, W. R. et al. Notch2 is a crucial regulator of self-renewal and tumorigenicity in human hepatocellular carcinoma cells. Oncol. Rep. 36 (1), 181–188 (2016).
Nguyen, P. H. et al. Characterization of biomarkers of tumorigenic and chemoresistant cancer stem cells in human gastric carcinoma. Clin. Cancer Res. 23 (6), 1586–1597 (2017).
Qiu, Y. et al. ALDH+/CD44 + cells in breast cancer are associated with worse prognosis and poor clinical outcome. Exp. Mol. Pathol. 100 (1), 145–150 (2016).
Ma, S. et al. Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations. Mol. Cancer Res. 6 (7), 1146–1153 (2008).
Lee, H. Y. & Hong, I. S. Targeting liver cancer stem cells: an alternative therapeutic approach for liver cancer. Cancers 12 (10), 2746 (2020).
Ma, X. L. et al. Sphere-forming culture enriches liver cancer stem cells and reveals Stearoyl-CoA desaturase 1 as a potential therapeutic target. BMC Cancer. 19, 1–12 (2019).
Visvader, J. E. & Lindeman, G. J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer. 8 (10), 755–768 (2008).
Singh, A. & Settleman, J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29 (34), 4741–4751 (2010).
Fung, S. W. et al. The ATP-binding cassette transporter ABCF1 is a hepatic oncofetal protein that promotes chemoresistance, EMT and cancer stemness in hepatocellular carcinoma. Cancer Lett. 457, 98–109 (2019).
Li, M. M. et al. Cancer stem cell-mediated therapeutic resistance in hepatocellular carcinoma. Hepatoma Res. 8, 36 (2022).
Mi, Y., Huang, Y. & Deng, J. The enhanced delivery of salinomycin to CD133 + ovarian cancer stem cells through CD133 antibody conjugation with Poly (lactic-co-glycolic acid)-poly (ethylene glycol) nanoparticles. Oncol. Lett. 15 (5), 6611–6621 (2018).
Akl, E. M., Abd-Rabou, A. A. & Hashim, A. F. Anti-colorectal cancer activity of constructed oleogels based on encapsulated bioactive Canola extract in lecithin for edible semisolid applications. Sci. Rep. 15 (1), 4945 (2025).
Fiorani, A. et al. Comparative performance of collagen nanofibers electrospun from different solvents and stabilized by different crosslinkers. J. Mater. Science: Mater. Med. 25, 2313–2321 (2014).
Jose, M. V., Thomas, V., Dean, D. R. & Nyairo, E. Fabrication and characterization of aligned nanofibrous plga/collagen blends as bone tissue scaffolds. Polymer 50 (15), 3778–3785 (2009).
Meng, Z. et al. Electrospinning of plga/gelatin randomly-oriented and aligned nanofibers as potential scaffold in tissue engineering. Mater. Sci. Engineering: C. 30 (8), 1204–1210 (2010).
Govender, T. et al. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J. Controlled Release. 57 (2), 171–185 (1999).
Hisaka, T. et al. Quercetin suppresses proliferation of liver cancer cell lines in vitro. Anticancer Res. 40 (8), 4695–4700 (2020).
Liu, Z. et al. Quercetin induces apoptosis and enhances gemcitabine therapeutic efficacy against gemcitabine-resistant cancer cells. Anti-Cancer Drugs. 31 (7), 684–692 (2020).
Asgharian, P. et al. Potential mechanisms of Quercetin in cancer prevention: focus on cellular and molecular targets. Cancer Cell Int. 22 (1), 257 (2022).
Mylonis, I., Lakka, A., Tsakalof, A. & Simos, G. The dietary flavonoid Kaempferol effectively inhibits HIF-1 activity and hepatoma cancer cell viability under hypoxic conditions. Biochem. Biophys. Res. Commun. 398 (1), 74–78 (2010).
Huang, W. W. et al. Kaempferol induces autophagy through AMPK and AKT signaling molecules and causes G2/M arrest via downregulation of CDK1/cyclin B in SK-HEP-1 human hepatic cancer cells. Int. J. Oncol. 42 (6), 2069–2077 (2013).
Almatroudi, A. et al. Effects and mechanisms of Kaempferol in the management of cancers through modulation of inflammation and signal transduction pathways. Int. J. Mol. Sci. 24 (10), 8630 (2023).
Huang, C. et al. Quercetin-induced degradation of RhoC suppresses hepatocellular carcinoma invasion and metastasis. Cancer Med. 13(4), e7082 (2024).
Hung, T. W. et al. Kaempferol inhibits the invasion and migration of renal cancer cells through the downregulation of AKT and FAK pathways. Int. J. Med. Sci. 14 (10), 984–993 (2017).
Ju, P. C. et al. Kaempferol inhibits the cell migration of human hepatocellular carcinoma cells by suppressing MMP-9 and Akt signaling. Environ. Toxicol. 36 (10), 1981–1989 (2021).
To, K. K. W. et al. Utilizing non-coding RNA-mediated regulation of ATP binding cassette (ABC) transporters to overcome multidrug resistance to cancer chemotherapy. Drug Resist. Updates. 73, 101058 (2024).
El-Ashmawy, N. E., Al-Ashmawy, G. M., Hamada, O. B. & Khedr, N. F. The role of ABCG2 in health and disease: linking cancer therapy resistance and other disorders. Life Sci. 360, 123245 (2025).
Martins-Gomes, C. & Silva, A. M. Natural products as a tool to modulate the activity and expression of multidrug resistance proteins of intestinal barrier. J. Xenobiotics. 13 (2), 172–192 (2023).
Xie, Z. Y. et al. Aspirin enhances the sensitivity of hepatocellular carcinoma side population cells to doxorubicin via miR-491/ABCG2. Biosci. Rep. 38 (6), BSR20180854 (2018).
Chen, Y. L. et al. ABCG2 overexpression confers poor outcomes in hepatocellular carcinoma of elderly patients. Anticancer Res. 36 (6), 2983–2988 (2016).
Zhang, G. et al. Expression of potential cancer stem cell marker ABCG2 is associated with malignant behaviors of hepatocellular carcinoma. Gastroenterology Research and Practice. 782581 (2013). (2013)(1).
Tsunedomi, R. et al. Elevated expression of RAB3B plays important roles in chemoresistance and metastatic potential of hepatoma cells. BMC Cancer. 22 (1), 260 (2022).
Namisaki, T. et al. Differential expression of drug uptake and efflux transporters in Japanese patients with hepatocellular carcinoma. Drug Metab. Dispos. 42 (12), 2033–2040 (2014).
Kalluri, R. & Weinberg, R. A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 119 (6), 1420–1428 (2009).
Lee, J. M., Dedhar, S., Kalluri, R. & Thompson, E. W. The epithelial–mesenchymal transition: new insights in signaling, development, and disease. J. Cell Biol. 172 (7), 973–981 (2006).
Zhan, X., Wang, F., Bi, Y. & Ji, B. Animal models of Gastrointestinal and liver diseases. Animal models of acute and chronic pancreatitis. Am. J. Physiology-Gastrointestinal Liver Physiol. 311 (3), G343–G355 (2016).
Patteson, A. E. et al. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 218 (12), 4079–4092 (2019).
Mitsiades, N. et al. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 61 (2), 577–581 (2001).
Strand, S. et al. Cleavage of CD95 by matrix metalloproteinase-7 induces apoptosis resistance in tumour cells. Oncogene 23 (20), 3732–3736 (2004).
Mao, J. et al. Roles of Wnt/β-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 5 (1), e1039–e1039 (2014).
Bao, S. et al. Glioma stem cells promote radioresistance by Preferential activation of the DNA damage response. Nature 444 (7120), 756–760 (2006).
Garg, H. et al. Survivin: a unique target for tumor therapy. Cancer Cell Int. 16, 1–14 (2016).
Pauklin, S. & Vallier, L. The cell-cycle state of stem cells determines cell fate propensity. Cell 155 (1), 135–147 (2013).
Sethi, G. et al. Apoptotic mechanisms of Quercetin in liver cancer: recent trends and advancements. Pharmaceutics 15 (2), 712 (2023).
Imran, M., Salehi, B., Sharifi-Rad, J. & Gondal, A. Kaempferol: A key emphasis to its anticancer potential. Molecules 24 (12), 2277 (2019).
Kim, S. H. & Choi, K. C. Anti-cancer effect and underlying mechanism (s) of kaempferol, a phytoestrogen, on the regulation of apoptosis in diverse cancer cell models. Toxicol. Res. 29, 229–234 (2013).
Li, X. et al. Quercetin alleviates pulmonary angiogenesis in a rat model of hepatopulmonary syndrome. Brazilian J. Med. Biol. Research 49(7) (2016).
Wu, L. et al. Quercetin shows anti-tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 8 (10), 4806–4820 (2019).
Lee, R. H. et al. Quercetin induces antiproliferative activity against human hepatocellular carcinoma (HepG2) cells by suppressing specificity protein 1 (Sp1). Drug Dev. Res. 76 (1), 9–16 (2015).
Luo, H. et al. Kaempferol inhibits angiogenesis and VEGF expression through both HIF dependent and independent pathways in human ovarian cancer cells. Nutr. Cancer. 61 (4), 554–563 (2009).
Zhu, G. et al. Kaempferol inhibits proliferation, migration, and invasion of liver cancer HepG2 cells by down-regulation of microRNA-21. Int. J. ImmunoPathol Pharmacol. 32, 2058738418814341 (2018).
Herrera, I. et al. Matrix metalloproteinase (MMP)-1 induces lung alveolar epithelial cell migration and proliferation, protects from apoptosis, and represses mitochondrial oxygen consumption. J. Biol. Chem. 288 (36), 25964–25975 (2013).
Yoneyama, M. S. et al. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur. J. Cell Biol. 93 (4), 157–169 (2014).
Colombo, M. et al. Kaempferol-loaded mucoadhesive nanoemulsion for intranasal administration reduces glioma growth in vitro. Int. J. Pharm. 543 (1–2), 214–223 (2018).
Dang, C. V. MYC on the path to cancer. Cell 149 (1), 22–35 (2012).
Yin, S. et al. Myc mediates cancer stem-like cells and EMT changes in triple negative breast cancers cells. PLoS One. 12 (8), e0183578 (2017).
Galardi, S. et al. Resetting cancer stem cell regulatory nodes upon MYC Inhibition. EMBO Rep. 17 (12), 1872–1889 (2016).
Cartwright, P. et al. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. (2005).
Liu, Z. et al. SOD2 is a C-myc target gene that promotes the migration and invasion of tongue squamous cell carcinoma involving cancer stem-like cells. Int. J. Biochem. Cell Biol. 60, 139–146 (2015).
Zhang, F. & Ma, C. Kaempferol suppresses human gastric cancer SNU-216 cell proliferation, promotes cell autophagy, but has no influence on cell apoptosis. Braz. J. Med. Biol. Res. 52 (2), e7843 (2019).
Shahbaz, M. et al. Anticancer and apoptosis inducing potential of Quercetin against a wide range of human malignancies. Int. J. Food Prop. 26 (1), 2590–2626 (2023).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This work received financial support from the Science, Technology and Innovation Funding Authority (STIFA), formally known as Science and Technology Development Fund (STDF), Egypt [Special targeted call (Health), Grant number 22930].
Author information
Authors and Affiliations
Contributions
(I) Conception and design: Hadeer A. Aglan, Ahmed A. Abd-Rabou, Hanaa H. Ahmed, Ghada H. Elsayed, Nadia S. Mahmoud (II) Nanoparticles preparation and characterization: Ahmed A. Abd-Rabou, (III) Isolation of CSCs and characterization as well as treatment with nanoparticles: Hadeer A. Aglan, Ghada H. Elsayed, Mohamed S. Kishta, Manar A. Elhinnawi, Nadia S. Mahmoud (IV) Data analysis and interpretation: Hadeer A. Aglan, Ahmed A. Abd-Rabou, Hanaa H. Ahmed, Ghada H. Elsayed, Nadia S. Mahmoud (V) Manuscript writing: Hadeer A. Aglan, Ahmed A. Abd-Rabou, Hanaa H. Ahmed, Ghada H. Elsayed, Nadia S. Mahmoud, (VI) Final approval of manuscript: All authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aglan, H.A., Abd-Rabou, A.A., Ahmed, H.H. et al. Role of CD133 antibody-conjugated nanocarrier in enhancing the targetability of hepatocellular carcinoma stem cells. Sci Rep 15, 30441 (2025). https://doi.org/10.1038/s41598-025-14435-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14435-9
Keywords
This article is cited by
-
The transforming role of wharton’s jelly mesenchymal stem cell-derived exosomes for diabetic foot ulcer healing: a randomized controlled clinical trial
Stem Cell Research & Therapy (2025)
-
Heightened anti-inflammatory and antioxidant effects of bone marrow stem cell-conditioned media with mono and doped TiO2 nanoparticles in Carrageenan-induced inflammation
Inflammopharmacology (2025)
