
Abstract
The zinc containing matrix metalloproteinase enzyme regulates a diverse array of biological processes in health and disease, including ADAM17 (a disintegrin and metalloproteinase domain 17) enzyme. Due to its large substrate profile, ADAM17 is known to regulate diverse pathways of inflammation and adaptive immunity. However, the role of ADAM17 in modulating the pathogenesis of type 2 allergic asthma is largely unknown. To determine the in vivo contribution of ADAM17 in house dust mite (HDM)-induced airway inflammation and adaptive immune response, we assessed the deletion of ADAM17 in mice conventional dendritic cells (ΔDC) and employed a complementary chemical biology approach using small-molecule novel ADAM17 inhibitor (2155-17). DC-specific ADAM17 ablation (ΔDC) suppressed type 2/ eosinophilic polarized HDM allergic responses and is protected from developing AHR. DC isolated from ΔDC mice showed a reduced state of metabolic activity, immune priming function and suppressed allergen-specific type 2 cell polarizations. Intranasal administration of 2155-17 protected WT mice against type2/ eosinophilic polarized HDM allergic responses. These concurrent results from two independent approaches identify a novel role for ADAM17 as an upstream site in airway inflammation. Furthermore, targeting ADAM17 with a selective small-molecule inhibitor might be harnessed as a potential drug target for type 2-high allergic asthma.
Similar content being viewed by others

Introduction
Allergic asthma is a chronic disease of the airways affecting more than 300 million people worldwide and continues to increase at a staggering rate by 50% over the next decade, with a rising healthcare economic burden of > 81.9 billion dollars1,2,3. In the clinic, 40 -70% of patients manifest type 2-high airway inflammation with elevated eosinophil infiltration in the lung tissues, a hallmark of allergic asthma4,5. The clinical phenotype of type2-high allergic asthma is a major medical problem with no known cure. Targeted therapies for type2-high/ eosinophilic asthma are quite limited despite the high prevalence of the disease6,7. Current pharmaceutical interventions to treat the disease are based on alleviating symptoms that emerge from airway inflammation. However, a generalized approach with an over-reliance on inhaled corticosteroid (ICS) and short-acting β agonist (SABA) therapy for an extended period without considering underlying inflammatory mechanisms during treatment selection to uncontrolled exacerbation of type2-high severe asthma7,8,9. Most recent studies of type 2 cytokine inhibition using monoclonal antibodies and type 2 biologic agents failed to mitigate the development of severe allergic and eosinophilic asthma and are discouraged10,11,12. Therefore, new strategies that provide fine-tuned therapeutic targeting of an upstream site in airway inflammation associated with allergic asthma are sorely needed.
ADAM17 (a disintegrin and metalloproteinase domain 17) is a membrane-anchored enzyme broadly expressed by immune cells, cleaving more than 80 different substrates in a cell-type and context-specific manner, thereby a promising drug target for the treatment of several inflammatory diseases13,14,15. Growing evidence from human and rodent studies suggests that ADAM17 activity is tightly regulated in the lung and is linked to the development of chronic lung diseases, including acute respiratory distress syndrome (ARDS), severe COPD, and fibrosis16. Temporal deletion of ADAM17 using tamoxifen-inducible cre-mediated recombination system in mice attenuated human neutrophil elastase-induced goblet cell metaplasia17. Interestingly, an increase in epidermal growth factor receptor (EGFR) signalling, a known substrate of ADAM17, is associated with airway thickening and exacerbation in asthmatics18,19,20. Overexpression of the EGFR dominant-negative form in mouse lung epithelium ameliorates HDM-induced airway smooth muscle thickening and airway hyperreactivity, suggesting a critical regulatory role of ADAM17 in lung inflammation21. The immune responses to inhaled allergens such as house dust mite (HDM) are orchestrated by antigen-presenting dendritic cells (DC), which are crucial in initiating and maintaining type 2/ eosinophilic airway inflammation22,23,24. Developing a robust allergen-specific type 2 immune response requires efficient recognition, activation, and migration of airway DC to the draining mediastinal lymph nodes (DLn) for T cell priming. More importantly, conditional removal of the conventional DC population in the lung abolishes all the cardinal features of asthma, including type2 cytokine-dependent airway eosinophilia, goblet cell hyperplasia, and bronchial hyperactivity, rendering airway DC at the interface of regulating inflammation and adaptive immune response22. Conventional airway DC (cDC), a major subset in the lungs, stems from clonogenic common DC progenitors in the bone marrow (BM), instigating allergic asthma25,26. Recently, we have identified that proliferation and differentiation of DC-restricted progenitors in the BM are coupled to intrinsic ADAM17 enzyme activity via MAPK-dependent pathway25. However, ADAM17 activity in airway DC regulating immune-priming function(s), and molecular mechanisms that consequently impact the adaptive immune response to inhaled allergen triggering eosinophilic/ type2-high asthma, are poorly understood.
To determine the in vivo contribution of ADAM17 activity in allergic asthma, we established conventional DC-specific ADAM17 transgenic mice (ΔDC) and bred floxed bearing ADAM17 with a Zbtb46 (cDC-restricted zinc finger transcription factor) cre-deleter strain27,28,29. We employed a clinically relevant murine model of experimental allergic asthma induced by repeated exposure to the aeroallergen HDM (D. pteronyssinus and D. farinae). Our results show that ΔDC mice are less susceptible to HDM-induced airway inflammation and type 2 immune response associated with allergic asthma. Using a complementary chemical biology approach, we developed a small-molecule ADAM17 inhibitor that acts via secondary site binding (non-zinc-binding inhibition) and found that intranasal administration of the inhibitor protected WT mice against type 2/eosinophilic polarized HDM allergic responses. These concurrent results from two independent approaches demonstrate a causal role for ADAM17 in type 2 airway inflammation and substantiate ADAM17 as a potential drug discovery target for allergic asthma.
Materials and methods
Ethical committee approval
All procedures were performed following relevant National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee of Auburn University, AL, USA. All mouse experiments were performed in accordance with relevant guidelines and regulations of the Institutional Animal Care and Use Committee, IACUC number 2020-3799 (2020–2022) and the study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org). Murine models of allergic asthma Low-endotoxin HDM (Dermatophagoides pteronyssinus and D. farinae extracts, < 15 EU/ mg. This study was designed to (i) examine ADAM17-mediated immunomodulatory function associated with type 2 airway inflammations and assess (ii) whether targeting ADAM17 by a novel small-molecule inhibitor in lungs suppresses asthma in mice. The impact of DC-specific ADAM17 ablation (ΔDC mice) and targeting ADAM17 with inhibitor (2155-17) was based on enumeration of (i) BAL and lung eosinophil and neutrophils by flow cytometry, (ii) pulmonary functions, (iii) lung histopathology, and (iv) Th2 cytokine productions by DLn. Adoptive transfer experiments of BMDC from fl/fl and ΔDC mice or 2155-17 inhibitor-treated were used to establish the in vivo cellular and molecular mechanism. All the repeats represent biological replication and involve the same experimental procedures utilizing different mice. All mice (anesthetized by 3% isoflurane) were euthanized using CO2 overdose at the end of experiments. Where there is a possibility of subjectivity, we have used double-blinded analyses to eliminate personal bias, especially for in vivo experiments. All samples from each group were analyzed in concert while comparing samples from different groups to prevent bias. All experiments were repeated at least thrice.
Animal care and study design
Six- ten weeks old, age- and sex-matched mice were used for all experiments in the study. ADAM17 floxed (ADAM17tm1.2Bbl/J), zDC-Cre (B6. Cg -Zbtb46 tm3.1(cre)Mnz/ J), BALB/C, and B6. Cg—Tg(TcraTcrb) 425Cbn/J mice on a C57BL/6 background were purchased from the Jackson Laboratory. ADAM17 floxed (fl/fl) and zDC-Cre mice were bred to generate conventional DC- specific conditional knockout (ΔDC mice), viable, fertile, and lacking any developmental abnormalities. Mice were housed and bred in a specific pathogen-free facility.
Measurement of airway hyperresponsiveness (AHR)
To measure AHR, fl/fl and ΔDC mice on day 16 post-challenge were mechanically ventilated and challenged with increasing concentrations of methacholine (Acetyl β-methyl choline, Sigma Aldrich, St. Louis, MO, USA). Briefly, mice were anesthetized and cannulated with a 19 -gauge beveled metal catheter connected to a ventilator (Elan RC Fine Pointe system; DSI, St Paul, MN, USA). AHR was measured at baseline and after linear dose-response with methacholine (0–25 mg/ ml) using a nebulizer. Airway resistance (RL; cm H2O per mL/s) and pulmonary compliance (Cdyn; cm H2O per mL/s) were recorded at 10 s intervals for 3 min, and mean ± SEM values were presented.
Lung histology
Lungs were fixed in 10% formalin for 24 h, dehydrated through gradient ethanol, and embedded in paraffin. Microtome Sections (5 µm thickness) of lungs were stained with hematoxylin and eosin and counterstained with periodic acid Schiff (PAS) to visualize goblet cells. All staining procedures were performed by the histology core facility at the College of Veterinary Medicine, Pathobiology Department, AU, Alabama, USA.
Ex vivo recall assay of DLn cells
Mediastinal lymph nodes (DLn) were isolated from mice, and single-cell suspensions were prepared using a sterile RPMI medium containing 10% FBS. DLn cells were counted and cultured (0.3 × 106 cells per ml) in round-bottom 96-well plates and restimulated with HDM (100 µg/ ml) for 96 h at 37 °C in RPMI medium containing 10% FBS. Cells were treated with ADAM17 inhibitor 2155–17 (50 µM) in selected experiments, and the concentrations of IL4, IL5, and IL13 in the supernatants were measured using sandwich ELISA (sensitivity limits: 4 pg/ ml; (ThermoFisher Scientific).
HDM-ELISA
Serum HDM-specific IgE and IgG1 levels were determined by ELISA, as previously described32,33. Biotinylated anti-mouse IgE or IgG1 (Pharmingen, San Jose, CA) and streptavidin-HRP (R&D Systems, Minneapolis, MN) secondary antibodies were used to determine bound HDM-specific antibodies.
Cell preparation and flow cytometry
Cells were isolated from the lungs, and bronchoalveolar lavage (BAL) was performed three times with 0.5 ml PBS as previously described24,25. Red blood cells were lysed with ACK buffer for 2 min at 4ºC, and BAL cells were re-suspended in 0.3 ml RPMI-1640 containing 10% FBS and stained for flow cytometry. BAL CCL24 levels were determined using quantitative ELISA kits from R& D Systems (Minneapolis, MN, USA). Lungs were isolated, cut into small pieces, and digested using type IV collagenase 1mgml-1 and DNase I (Worthington, Lakewood, NJ; 0.1 mg ml-1) at 37 ºC for 30 min. Cells were incubated with Fc BlockTM (BD Pharmingen, San Jose, CA) and surface stained at 4 ºC for 10 min. Cells were first stained with Live/ dead Fixable Violet Staining Kit (Invitrogen) and eosinophils (CD45 + CD11b + SiglecF + Ly6G-) and neutrophils (CD45 + CD11b + SiglecF-Ly6G +) in BAL and lung were identified using the following antibodies: rat anti-mouse CD45 efluor 450 (clone 30-F11), CD11b-APCCy7 (clone M1/70) Ly6G-APC (clone 1A8) and Siglec F-PE-Texas Red (clone 1RNM44N). Lung DC population were gated on Siglec F-/ CD11c + /MHC-IIhi /SSClo, and DC subsets were further enumerated with SIRPα-PE (clone P84), CD103- PerCP-efluor 710 (clone 2E-7), and PDCA1-PE-efluor 610 (clone eBio 927). Surface DC-specific activation markers were identified in DLn with rat anti-mouse CD11c-APC-eFluor780 (clone N418), I-A/I-E (MHC-II)-eFluor450 (clone M5/114.15.2), CD11b-PE-TexasRed (M1/70.15), Siglec F-eFluor660 (1RNM44N), CD86-PE (clone, PO3), CD80-PE-Cy5 (clone, 1610A1), CCR7- APC (clone, 4B12), and CD40-FITC (clone, 323). To identify BAL cytokines, cells were first stained with surface antigens against rat anti-mouse CD45 efluor 450 (clone 30-F11), CD3-Alexa Fluor 647 (clone 17-A2), and CD4-APC-eFluor 780 (clone GK1.5). Fixed cells (Fixation Buffer; eBiosciences) were re-suspended and reacted to monoclonal antibody cocktails of rat anti- mouse IL-4 PE-Cyanine7 (11B11), IL-5 PE (TRFK5), IL-13 Alexa Fluor 488 (eBio13A), IL-17A eFluor 450 (eBio17B7) in Perm/Wash buffer (50 μl) for 30 min at room temperature. Viable CD3 + / CD4 + cytokine + cells were quantified using FMO (fluorescence minus one) as controls. To quantify Tregs, DLn cells were first stained with surface markers for CD3-Alexa Fluor 647 (clone 17-A2), CD4-FITC (clone GK1.5), and CD25-PE-Cy7 (clone PC61.5) and fixed and permeabilized using Foxp3 staining buffer (eBiosciences) and reacted to Foxp3-PE antibody (clone NRRF-30). In separate experiments, lung and spleen cells were identified with DC surface markers and fixed/ permeabilized with anti-ADAM17 monoclonal antibody (Abcam, Waltham, MA) labeled with APC for intracellular staining. Data were acquired on an LSRII (BD Biosciences, USA) equipped with 407, 488, and 633 LASER lines using DIVA 8 software and analyzed with the Flow Jo software version 10.7.1 (Treestar, San Carlos, CA). Cellular debris was excluded using a forward light scatter/side scatter plot.
Preparation of bone marrow-derived DC, cell purifications, and adoptive DC transfer model of asthma
Cells were isolated from femur bones of euthanized fl/fl and ΔDC mice and cultured at a density of 1 × 106 cells/ ml in differentiation media (Iscove modified Dulbecco medium (Gibco/Life Technologies, CA) containing 10% heat-inactivated FCS, penicillin–streptomycin (100 U/ ml), L- glutamine (2 mM), 2-mercaptoethanol (50 µM), recombinant mouse granulocytes macrophage colony-stimulating factor (20 ng/ ml) and recombinant mouse IL-4 (10 ng/ ml, Peprotech). On day six, non-adherent cells were collected and cultured at a density of 2 × 106 cells/ well in differentiation media and pulsed with HDM (100 μg/ ml) for 16 h at 37 ºC before ADAM17 activity measurements and XFp assay. For adoptive transfer experiments, HDM- (100 µg/ ml) or PBS- pulsed BMDC from fl/fl and ΔDC mice or 2155–17-treated (50 μM) or untreated BMDC were enriched with EasySep Mouse Pan-DC Enrichment Kit according to the manufacturer’s instructions (StemCell Technologies). Negatively sorted live CD11c + BMDC (0.75 × 105 cells) were adoptively transferred in 40 µl of PBS through intranasal administration on day 0 to recipient naïve WT B6 mice. Recipient naïve mice received daily HDM challenges (50 µg, i.n.) on days 9, 11, 13, 15 and were sacrificed on day16 as previously described24,30. Two independent experiments were performed for BMDC adoptive transfer model.
Measurements of antigen uptake, migration, and presentation
Fl/fl and ΔDC mice received Alexa fluorophore 647-labeled OVA (i.n.; 100 µg per sensitization) dissolved in 40 µl of PBS. Antigen uptake by CD11b + lung DC and migration of DC to DLn were enumerated as SiglecF- CD11c + MHCIIhi CD11b + AF647-OVA + cells by flow cytometry. To determine the antigen presentation capability of DC, antigen-specific T-cell proliferation was assessed ex vivo using CFSE-labeled splenic CD4 + T cells isolated from naïve transgenic OTII mice (B6.Cg-Tg(Tcra Tcrb)425Cbn/J) (Jackson Laboratories, Bar Harbor, ME) co-cultured with DC. The mice express a transgenic MHCII-restricted TCR and recognize the OVA peptide antigen. Splenic naive CD4 + T cells were purified and enriched (EasySep Mouse CD4 + T Cell Isolation Kit; Stem cells, Vancouver, CA) and were labeled with CFDA-SE (5 µM; carboxyfluorescein diacetate succinimidyl ester; Cayman Chemical, MI, USA) in PBS for 20 min at 37 °C. BMDC from fl/fl and ΔDC mice and 2155-17 (50 μM)- or ADAM 10-, Meprin- (20 μM)- treated, or untreated cells were pulsed overnight with OVA323-339 peptide (5 ng/ ml; AnaSpec, Fremont, CA) or PBS. OVA peptide-specific CD4 + OT-II (1 × 105) cells were cocultured with BMDC (2 × 104) for 4 days, and T cell proliferation was quantified by the CFSE dye dilution method. The FlowJo Proliferation Platform analyzed gated CD4 + T-cells as percent divided. The IL4 and IL13 released into the culture medium were measured using ELISA. Additionally, cells were stained with CD3-Alexa Fluor 647 (clone 17-A2) CD4-APC-eFluor 780 (clone GK1.5) before incubation with GATA3 eFluor 450 (clone TWAJ) for flow cytometry.
Seahorse assay
HDM-pulsed BMDC treated with 2155-17 (50 μM) or obtained from fl/fl, and ΔDC mice were utilized for oxygen consumption rate (OCR) measurements using an XFp extracellular flux analyzer (Seahorse Bioscience). Briefly, BMDC (120,000 cells per well; XFp mini-culture plates) were pulsed with HDM (100 µg/ ml) overnight before the assay in IMDM medium containing 5% FBS at 37 °C as described previously30. Mitochondrial stress tests were performed by sequential injections of oligomycin (1 mM, mitochondrial ATP synthase inhibitor), carbonyl cyanide 4- (trifluoromethoxy) phenylhydrazone (FCCP, 2 mM, uncoupler of OXPHOS from ATP synthesis), and antimycin A/rotenone (0.5 mM, ETC inhibitor) in the port. The effects on OCR were recorded 3 times every 5 min interval, and data were analyzed using Wave software 2.6.1 (Seahorse Bioscience) after normalization with total protein. Spare respiratory capacity (SRC) was calculated as the difference between basal OCR values and maximal OCR values achieved after FCCP uncoupling.
Real-time PCR
RNA was isolated from lung, CD11c + lung DC (mouse cell enrichment kit; Stem cells, Vancouver, CA), and cultured BMDC of fl/fl and ΔDC mice using trizol reagent (Life Technologies, Grand Island, NY, USA). cDNA was synthesized by a High Capacity RNA-to- cDNA kit (Applied Biosystems). qPCR for ADAM17 (Applied Biosystems; Mm00456428_m1) and β-actin (Mm02619580_g1) were performed using Taqman gene expression assay (Life Technologies). Results were analysed using 2 −ΔΔCt method and expressed relative to β-actin.
ADAM17 activity and MMP assays
ADAM17 activity (SensoLyteTM 520) was measured in BMDC from fl/fl and ΔDC mice according to the manufacturer’s protocol (Anaspec, San Jose, CA) based on fluorescence resonance energy transfer (FRET). The fluorescent signal was proportional to the reaction and recorded at Ex/Em = 480 nm/ 520 nm for 60 min using the SPECTRAmax fluorometer. In separate assays, ADAM17 inhibitor 2155-17 and pharmacological control (marimastat) were added to solid bottom black plates (Nunc, cat# 264705) using a 384-pin tool device (V&P Scientific, San Diego) that contains 5 μL of 2 × enzyme solution (5 nM) in assay buffer (50 mM Tricine, 50 mM NaCl, 10 mM CaCl2, 0.05% Brij-35, pH 7.5). After 60 min incubation at room temperature, the reactions were started by adding 5 μL of 2 × solutions of MMP substrate (R&D Systems cat#: ES010, 20 μM). Reactions were incubated for 1 h at room temperature, after which the fluorescence was measured using the Synergy H1 multimode microplate reader (Biotek Instruments) (λ excitation = 324 nm, λ emission = 390 nm).
Synthesis, biochemical and cell-based assays to test potency/selectivity/toxicity of ADAM17 inhibitor
Synthesis of diketopiperazine pyrrolidine-based ADAM17 inhibitors has been reported previously31. Briefly, 2155-17 was synthesized from the peptide precursor using standard solid-phase amino acid and carboxylic acid couplings. The amide bonds are then reduced with borane providing a polyamine treated with oxalyldiimidazole to form two diketopiperazine rings. After cyclization, the product is removed from the resin and purified utilizing RP-HPLC. The procedure is amenable to multi-gram scale synthesis. The final compound is purified using preparative HPLC with a dual pump Shimadzu LC-20AB system equipped with a Luna C18 preparative column (21.5 × 150 mm, 5 microns) at λ = 214 nm, with a mobile phase of (A) H2O (+ 0.1% formic acid)/(B) acetonitrile (ACN) (+ 0.1% formic acid) at a flow rate of 13 mL/min. The purity (> 95%) and structure were confirmed by HPLC-LC/MS and 1H and 13C NMR, respectively. Inhibition Kinetics: Glycosylated substrate and ADAM17 working solutions were prepared in buffer containing 10 mM HEPES, 0.001% Brij-35, pH 7.5. All reactions were conducted in 384- black polystyrene well plates (Nunc, cat# 264706). Determinations of inhibition constants and modalities were performed by incubating the range of glycosylated substrate concentrations (1– 75 µM) with 2.5 nM ADAM17 at room temperature in the presence of varying concentrations of 2155-17. Fluorescence was measured as described above. Rates of hydrolysis were obtained from plots of fluorescence versus time, using data points from only the linear portion of the hydrolysis curve. All kinetic parameters were calculated using GraphPad Prism version 9.01 (GraphPad Software, Inc., La Jolla, CA). Ki and Ki’ values were determined by non-linear regression (hyperbolic equation) analysis using the mixed inhibition model, which allows for the simultaneous determination of the mechanism of inhibition31,32. The mechanism of inhibition was determined using the “alpha” parameter derived from a mixed model inhibition by GraphPad Prism. Lineweaver–Burk plots additionally confirmed the mechanism of inhibition.
Statistical analysis
The statistical power was obtained using 3–4 mice per group to characterize unbiased results from AHR measurements, flow cytometry analysis of BAL and lung cellularity, lung immune response, DLn recall experiments, coculture studies, and in vitro assays. The statistical analysis for sample group size to provide a power of 0.9 (when α = 0.5%) was calculated using the SAS program. All results are expressed as means + /− SEM, and P-value < 0.05 was considered significant. Statistical significance was obtained using Prism 7.05 software, either by an unpaired t-test for normally distributed data or a Mann–Whitney test for non-normally distributed data. Comparisons between more than one group were analyzed using a one-way ANOVA with Tukey’s multiple comparisons test for all experiments, except ADAM17 lead testing of metzincin selectivity, mechanism of inhibition, and toxicity assays (Fig. 4), which were statistically analyzed using Dunett’s post hoc test.

ADAM17 ablation in cDC reduces type 2/ eosinophilic polarized HDM allergic responses. (A) Experimental protocol for treating HDM-induced asthma exacerbation. A cohort of ADAM17 floxed (fl/fl) or ADAM17 fl/fl; Zbtb46-Cre (ΔDC) mice were sensitized with two intraperitoneal injections of HDM (100 μg) or PBS (with 40 mg/ ml alum) on days 0 and 4 and challenged on days 8, 10, 12, and 14 employing intranasal administration of HDM (50 μg) before endpoint analysis on day 16. (B) Pseudocolor plots show representative frequencies and (C) absolute numbers of BAL (left) and lung (right) eosinophils (CD11b + Siglec-F +) and neutrophils (CD11b + Ly-6G +) (n = 3 to 7 mice per group). Data are representative of three independent experiments. Bar chart shows (D) BAL CCL24 levels and (E) serum HDM-specific IgG1 and IgE levels from asthmatic mice. (F) Airway resistance (RL; left) and dynamic compliance (Cdyn; right) to inhaled methacholine were determined on day 16 as described in Materials and Methods. (G). Cytokine production by lung draining lymph nodes (DLn) from fl/fl or ΔDC asthmatic mice that had been re-stimulated ex vivo with HDM (100 μg/ml) for 4 days (n = 3 to 7 mice per group). Data are representative of three independent experiments. (H) Representative histologic images of lung sections stained with H&E and PAS. Scale bars, 100 μm for × 100 and 50 μm for × 400 images. Graphs represent the mean, with error bars indicating SEM. *p < 0.05, fl/fl versus ΔDC, determined by unpaired Student’s t-test (d–f) or one-way ANOVA with Tukey’s multiple comparison test (c) and (g). OD450, optical density at 450 nm.
Results
cDC-specific deletion of ADAM17 prevented HDM-induced asthma exacerbation
Antigen-presenting conventional lung DC (cDC) determines the initiation and propagation of airway inflammation in allergic asthma24,33. Previously, we have identified that ADAM17 enzyme activity is required for the proliferation and differentiation of DC-restricted progenitors25. Here, we hypothesized that ADAM17 enzyme activity in cDC is a critical determinant in Th2 cell- mediated immunity to inhaled aeroallergen house dust mite (HDM) allergic responses. We bred mice with floxed ADAM17 with a Zbtb46 (a zinc finger transcription factor evolutionarily conserved and expressed explicitly by conventional DC, but not monocytes or other immune cells; Cre deleter strain (ΔDC mice)14. There were no differences in gross pathology, litter size, weights, viability, and fertility between fl/fl and ΔDC mice strains. To study the role of ADAM17 in cDC-mediated adaptive immunity and the development of allergic Th2/eosinophilic airway inflammation, we employed a clinically relevant murine model of experimental asthma induced by repeated exposure to HDM (Dermatophagoides pteronyssinus and D farinae extracts from Greer Laboratories) (Fig. 1A). Twenty-four hours after the last allergen challenge, HDM stimulation markedly induced ADAM17 expression in the lung and restricted to lung CD11c + DC population or HDM-pulsed bone-marrow-derived DC (BMDC) from fl/fl mice, which is significantly attenuated in ΔDC mice (Supplementary Figure S1A,B). Furthermore, by flow cytometry, we confirmed that ADAM17 protein expression is restricted to the CD11b + conventional DC in the fl/fl lung and is absent from the lung of ΔDC mice (Supplementary Figure S1C). Our results demonstrate that asthmatic ΔDC mice were completely protected from HDM-induced asthma exacerbation as manifested by reduced frequencies (Fig. 1B) and absolute number (Fig. 1C) of CD11b + Siglec F + eosinophils in the BAL and lung as compared to fl/fl mice. However, the numbers of neutrophils in BAL and lung from both genotypes were not detected. As shown in Fig. 1D,E, eosinophil recruiting CCL24 (C–C chemokine) in the BAL and serum HDM-specific immunoglobulin G1 (IgG1) and IgE levels were significantly attenuated in the ΔDC asthmatic mice compared with the fl/fl mice. The analysis of lung function showed improved lung function with decreased airway resistance and increased pulmonary compliance in ΔDC asthmatic mice compared to HDM treated fl/fl mice (Fig. 1F). Furthermore, experiments were next conducted to assess whether the deletion of ADAM17 in DC impacted the proportion of DC subtypes. The frequencies of major CD11b + SIRPα + or CD103 + SIRPα- conventional lung DC population and PDCA1 + plasmacytoid DC population were not significantly different in the lungs and draining mediastinal lymph nodes (DLn), indicating that DC development was not significantly impaired in fl/fl and ΔDC asthmatic mice (Supplementary Figure S1D). We found no differences in Tregs percentage in DLn of HDM-challenged fl/fl and ΔDC. Surface expression of the costimulatory molecules (CD80, CD86, and CD40) was significantly reduced on CD11b + conventional DCs in the lungs of HDM-challenged ΔDC mice compared with fl/fl mice ((Supplementary Figure S1E,F). However, there was no difference in CCR7 expression by CD11b + conventional DCs in the fl/fl and ΔDC lungs. Furthermore, examining HDM-specific memory Th2 cells in DLn by ex vivo recall studies demonstrated a marked decline in IL4, IL5, and IL13 production from ΔDC mice as compared with fl/fl control mice (Fig. 1G). Last, lung histopathology showed reduced peribronchial inflammatory cell infiltrates and mucous cell metaplasia in HDM-challenged ΔDC mice as compared with fl/fl control mice (Fig. 1H). Altogether, these data indicate that ΔDC lungs immunized with HDM were protected from developing airway hyperresponsiveness (AHR) and airway inflammation, suggesting the regulatory role of cDC-specific ADAM17 activity in mediating allergic eosinophilic asthma.
ADAM17 activity in DC contributes to the induction of airway inflammation and asthma
Because cDC-restricted deletion of ADAM17 in ΔDC mice has defects in mounting Th2/ eosinophilic airway inflammation, we next assessed whether DC from ΔDC mice has an impairment that limits their ability to induce allergic sensitization and T cell-mediated immune responses to inhaled HDM. Bone-marrow-derived dendritic cells (BMDC) were isolated from fl/fl and ΔDC mice and pulsed with HDM. As shown in Fig. 2A, ADAM17 activity was induced in HDM-pulsed BMDC isolated from fl/fl mice and is absent in ΔDC BMDC. Since DC exhibits metabolic switch upon allergen stimulation that influences immune-priming functions30,34, we next measured the oxygen consumption rate (OCR) as an indicator of mitochondrial OXPHOS in BMDC isolated from fl/fl and ΔDC mice. In the presence of HDM, BMDC from fl/fl mice showed typical OCR changes in response to sequential inhibition of mitochondrial ATP synthase by oligomycin (oligo), uncoupling of OXPHOS from ATP synthesis with FCCP, ETC inhibition by antimycin/ rotenone (Fig. 2B). In contrast, ΔDC BMDC were only slightly responsive to these drugs as indicated by the reduced frequency of spare respiratory capacity (SRC) and coupling efficiency, suggesting metabolic adaptations of DC are coupled to ADAM17 activity.

HDM-pulsed CD11c + BMDCs from Zbtb46-specific ADAM17 knockout mice (ΔDC) are metabolically less active and have impaired ability to drive type 2/ eosinophilic airway inflammation. (A) Kinetics of ADAM17 enzymatic activity analysis using ADAM17-specific FRET peptide cleavage by HDM-pulsed (100 μg/ml) fl/fl or ΔDC BMDC. (B) Extracellular flux analysis of HDM-pulsed BMDC isolated from fl/fl (black) or ΔDC (red) mice. A metabolic stress test was performed, and kinetic oxygen consumption rate (OCR) was determined in response to sequential oligomycin (oligo, 1 mM), carbonyl cyanide4-(trifluoromethoxy) phenylhydrazone (FCCP, 2 mM), and antimycin A/ rotenone (0.5 mM) treatments after basal measurements. Arrowhead indicates drug injection point. Bar graph shows the spare respiratory capacity (SRC; left) calculated as the difference between basal OCR values and maximal OCR values achieved after FCCP uncoupling, and coupling efficiency percentage (ATP production rate/ basal respiration rate × 100; right). Data are expressed as mean + /− SEM, (n = 4; *p < 0.05, fl/fl HDM versus ΔDC HDM; unpaired Student’s t-test. (C) Experimental protocol for DC-driven asthma model. CD11c + BMDC from fl/fl or ΔDC mice were HDM-pulsed (100 μg/ml) or treated with PBS for 16 h and were adoptively transferred to naïve WT mice (0.75 × 105 cells in 40 μl sterile saline, i.n. administrations). Recipient WT mice received HDM challenges (50 μg; i.n.) on days 9, 11, 13, and 15 before harvest on day 16. (D) Absolute numbers of total inflammatory cells, eosinophils, and neutrophils in BAL (left) and lung (right) in recipient WT mice. Bar graph represents (E) serum level of HDM-specific IgG1 and IgE and (F) frequencies of CD4 + cytokine + cells in BAL. Data are expressed as mean + / − SEM and represent one of the two adoptive transfer experiments (n = 3 to 7 mice per group). (G) IL4, IL5, and IL13 in culture supernatants of DLn cells were re-stimulated ex vivo with HDM (100 μg/ ml). (H) Representative H&E and PAS staining of lung sections from WT recipient mice receiving HDM or PBS-pulsed BMDC from fl/fl or ΔDC donor mice. Scale bars, 100 μm for × 100 and 50 μm for × 400 images. Graphs represent the mean, with error bars indicating SEM. *p < 0.05, fl/fl HDM-WT versus ΔDC HDM-WT as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Next, to confirm that DCs explicitly mediated the suppressed HDM-induced type 2 /eosinophilic airway inflammatory response in ΔDC mice, we transferred HDM-pulsed CD11c + BMDC adoptively to naïve WT mice that subsequently received multiple intranasal HDM challenges to induce allergic airway inflammation (Fig. 2C). Absolute number of BAL and lung cells, predominantly composed of eosinophils, was significantly reduced in the WT recipient mice receiving sensitized ΔDC BMDC compared with fl/fl BMDC (Fig. 2D). Serum levels of HDM- specific IgG1 and IgE were markedly reduced in WT recipient mice that received the adoptive transfer of CD11c + BMDC from ΔDC donor mice compared with WT recipients of CD11c + BMDC from fl/fl donor mice (Fig. 2E). In addition, the frequencies of CD4 + IL5 + and CD4 + IL13 + cells in BAL from WT recipient mice that received CD11c + BMDC from ΔDC mice were significantly reduced; however, CD4 + IL4 + and CD4 + IL17A + cells remain unchanged as compared to WT recipient mice that received sensitized BMDC from fl/fl mice (Fig. 2F). HDM restimulation of cultures of DLn from WT asthmatic mice that received sensitized BMDC from ΔDC donor mice showed a significant reduction in the productions of Th2 cytokines (IL4, IL5, and IL13) as compared to WT asthmatic mice receiving sensitized BMDC from fl/fl donor mice (Fig. 2G). Last, lung histology revealed decreases in peribronchial and perivascular inflammatory cell infiltrates in recipient WT mice that received the adoptive transfer of BMDC from ΔDC mice compared with WT recipients of BMDC from fl/fl donor mice (Fig. 2H). As such, these results collectively demonstrate that ADAM17-deficient DC has an impaired ability to mount HDM-mediated allergic sensitization and type 2 airway inflammations, thereby restating ADAM17 activity in regulating DC-intrinsic function(s) and allergic eosinophilic asthma.
Lung DC from fl/fl and ADAM17-deficient mice differ in their antigen-presenting capability
Lung DC are primary antigen-presenting cells necessary for type 2 polarized HDM allergic responses. Thus we tested whether ADAM17 activity in DCs modifies antigen-specific T cell proliferation and type 2 cytokine production as a possible mechanism of attenuated type 2/ eosinophilic airway inflammation in ΔDC mice to HDM. Coculture experiments were conducted that comprised splenic CD4 + T cells from OTII transgenic mice expressing MHCII-restricted α- chain and β-chain TCR that explicitly recognize the OVA323-339 peptide. In the presence of OVA323-339 peptide-pulsed CD11c + BMDC from fl/fl mice, there was an increase in proliferation of OTII cells, whereas BMDC from ΔDC mice failed to mediate T cell proliferation (Fig. 3A).

ADAM17 induction is required for DC priming and antigen-specific Th2 polarization. BMDC from fl/fl and ΔDC mice were pulsed with the OVA323-339 peptide (1 μg/ ml) and incubated at a 1:5 ratio with CFSE-labeled splenic CD4 + OTII cells for 4 days. (A) Bar graph shows the OVA-specific proliferation of OTII cells (each circle represents individual mice). (B) Pseudocolor plots show representative flow cytometric data (left) and frequencies of CD4 + GATA3 + OTII cells (right) in coculture and (C) Th2 cytokines released by coculture of OVA323-339 peptide-pulsed BMDCs and CFSE-labeled OTII cells (n = 6). Data are representative of three independent experiments and represent the mean with error bars indicating SD. *p < 0.05, fl/fl OVA323-339 versus ΔDC OVA323-339 as determined by one-way ANOVA with Tukey’s multiple comparison test. (D) Percentage of OVA-AF647 + CD11b + DC (gated on Siglec-F- CD11c + MHCIIhi population) in the lung and draining lymph node (DLn) of fl/fl or ΔDC mice 72 h after the allergen administration. Bar graph represents the mean + / − SEM from two independent experiments (n = 6 per group, *p < 0.05 unpaired Student’s t-test).
Moreover, the frequency of CD4 + GATA3 + cells (Th2-specific transcription factor) was markedly reduced in the presence of OVA323-339 peptide-pulsed ΔDC BMDC compared to the fl/fl BMDC after four days of ex vivo culture (Fig. 3B). Culture supernatants of BMDC from ΔDC mice showed marked reduction in Th2 cytokine production (IL13 and IL4) compared with fl/fl BMDC after ex vivo stimulation to OVA323-329 peptide, suggesting the impaired ability of ΔDC mice to present antigen to CD4 + T cells and thereby suppressed effector type 2 inflammatory response (Fig. 3C). Next, to assess the functional capacity of CD11b + conventional lung DC to capture antigen and transport allergen-derived antigens into the DLn, fluorescent (AF647)- labeled OVA was administered to the lungs of naïve fl/fl and ΔDC mice. Thus, we inoculated (i.n.) OVA-647 (100 μg per mouse) during the sensitization phase and frequencies of OVA- bearing AF647 + CD11b + DC in lung and DLn were analyzed and enumerated at 72 h by flow cytometry. As shown in Fig. 3D, the frequency of OVA-bearing, Alexa 647 + CD11b + from fl/fl and ΔDC mice were identical in lungs (49.8% ± 3.7% vs. 40.9% ± 2.6%) and DLn (43.6% ± 1.9% vs. 38.1% ± 1.7%). Thus the differences in Th2 responses between fl/fl and ΔDC mice after allergen sensitization were not due to differences in the capacity of CD11b + DC to capture or subsequent transport of allergen-derived antigen to draining DLn but to their impaired ability in antigen presentation to T cells.
Generation and initial optimization of selective non-zinc-binding exosite inhibitors of ADAM17
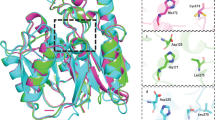
Multiple drug discovery programs targeting ADAM17 have been abandoned due to the toxicity of drug candidates35,36. It is widely accepted in the zinc metalloprotease field that the toxicity of ADAM17 inhibitors is due to the broad inhibition profile against members of the metzincin family of enzymes (ADAM, MMP, and ADAMTS)37. Most of the ADAM17 inhibitors developed to date feature Zn-binding moieties that target the active site Zn, which explains the off-target toxicity of Zn-binding inhibitors. We developed a novel diketopiperazine pyrrolidine-based ADAM17- selective small-molecule inhibitor (2155-17) that acts via secondary site binding (non-zinc- binding inhibition) from the parallel screening of mixture libraries against ADAM10 and 17 identified library 1344 (Fig. 4A). Previously, we have demonstrated that 2155-17 do not act via binding Zn of an active site but rather by binding to the secondary binding site within the ADAM17 moiety38. Based on the “scaffold ranking” screen results, we prioritized library 1344 for the full positional scanning deconvolution. The positional scan results identified several functional group substitutions that exhibited desired activity and selectivity. A few substitutions were selected at each variable position providing target compounds, which were synthesized and tested against the target enzyme (ADAM17) and counter targets (ADAM10, MMPs) in dose- response experiments. All compounds exhibited activity (single-digit µM) against ADAM17 with high selectivity over the counter targets (Fig. 4B), including 2155-17 (active in ex vivo and in vivo) identified as a lead. Mechanistic studies performed on 2155-17 showed that it is a non-competitive inhibitor of ADAM17 (Fig. 4C) that binds to an unknown site outside of the active site of ADAM17 and does not bind zinc of an active site (Fig. 4D), which explains its selectivity. The lead 2155-17 exhibited enzyme and substrate selectivity in a cell- based setting31,38. To pre-empt potential toxicity problems, we tested 2155-17 with mouse oligodendrocyte progenitor cells (OPCs). As shown in Fig. 4E,F, the lead 2155-17 increased OPCs differentiation and is independent of apoptotic cell death (Fig. 4G) in a dose-dependent manner, suggesting that the compound has no toxic effects. Collectively, these data demonstrate that our novel ADAM17 inhibitor 2155-17 has a unique enzyme, substrate, and off- target selectivity profile due to its innovative exosite-binding mechanism of inhibition and can be used in vivo.

ADAM17 lead testing of metzincin selectivity, mechanism of inhibition, and toxicity. (A) Structure of ADAM17 lead 2155-17, (B) dose-response study of ADAM17 lead against a panel of metzincins. (C) Non-linear regression analysis shows the best fit to a non-competitive inhibition mechanism. (D) Lineweaver-Burke plot confirms the non-competitive inhibition mechanism of ADAM17 lead. The arrow points to the lines of best-fit crossing above the X-axis. (E) Oligodendrocyte precursor cells (OPCs) were treated with or without ADAM17 inhibitor 2155-17, and (F) quantification of images showing a dose-dependent increase in OPCs differentiation in the presence of 2155-17. (G) Quantification of pyknotic cells with different dosages of 2155-17. One-way analysis of variance (ANOVA) was used, followed by a Dunnett post hoc test. The data shown are the mean + / − SEM, n = 6; **p < 0.01, ns not significant. T3 thyroid hormone (10 ng/ ml); DMSO (0.1%).
2155-17-mediated pharmacological inhibition of ADAM17 impairs DC immune priming functions, thereby attenuating type 2/ eosinophilic polarized HDM allergic responses
Since we found metabolic adaptations are coupled to ADAM17 activity in DC that affects immune-primming functions, experiments were next performed with BMDC treated with ADAM17 inhibitor 2155-17 for mitochondrial respiratory activity. Concordant with the BMDC results from ΔDC mice, there was a significant reduction in HDM-induced OCR and corresponding spare respiratory capacity (SRC) with 2155-17 treatment relative to controls (Fig. 5A,B). Additionally, our results using CD4 + T cells from mice expressing the MHCII-restricted T cell receptor that recognizes the OVA323-339 peptide showed that BMDCs treated with 2155-17 have a reduced ability to induce T cell proliferation (Fig. 5C), Th2 cytokine IL13 production (Fig. 5D), and GATA3 expression (Fig. 5E,F) as compared with untreated control. Moreover, BMDC treated with other metzincin inhibitors such as ADAM10 and Meprin inhibitors could not inhibit Ag-specific T cell proliferation, GATA3 expression, and IL13 release, indicating the specificity of 2155-17 in DC priming capacity. Because ΔDC mice have defects in mounting Th2/eosinophilic airway inflammation, we next assess whether sensitization of naïve mice with 2155-17-treated CD11c + BMDC ex vivo has a similar effect in response to HDM (Fig. 6A). Recipients of adoptively transferred HDM-pulsed BMDC that had been treated with 2155-17 had a significant decrease in the number of BAL inflammatory cells as compared with recipients of HDM-pulsed BMDC that had been treated with vehicle alone (Fig. 6B). Moreover, the surrogate chemokine (CCL24) of BAL eosinophilia, serum allergen-specific IgG1, and IgE levels were declined in recipient WT mice receiving 2155-17-treated BMDC relative to WT mice receiving untreated BMDC (Fig. 6C,D). Consistent with this, DLn cells from recipients of 2155-17 treated HDM-pulsed BMDC had a reduced ability to produce the Th2 cytokines IL4, IL5, and IL13 compared with recipients of HDM-pulsed WT BMDC treated with vehicle alone (Fig. 6E). Likewise, adoptive transfer of 2155-17-treated sensitized BMDC to naïve WT mice showed a reduction in peribronchial inflammatory cell infiltrates and associated mucous cell metaplasia compared with recipients of HDM-pulsed BMDC that had been untreated as evidenced by histology (Fig. 6E,F). Indeed, the protective effects of 2155-17-mediated pharmacological inhibition of ADAM17 in DC immune-priming capacity and parallel attenuating type 2 /eosinophilic polarized HDM allergic responses were indistinguishable from benchmarks in allergic ΔDC mice, strongly supporting the causal role of ADAM17 in type 2 airway inflammations and asthma.

Effect of ADAM17 inhibitor (2155-17) treatment on the capacity of DC to activate and polarize antigen-specific T cells in vitro. (A) Representative oxygen consumption rate (OCR) trace from HDM-pulsed BMDC in presence or absence of ADAM 17 inhibitor 2155-17 (50 μM). A metabolic stress test was performed, and spare respiratory capacity (SRC, right) was calculated as the difference between basal OCR values and maximal OCR values achieved after FCCP uncoupling. Arrowhead indicates drug injection point. (B) Bar graph represents mean + / − SD of SRC ratio, defined as SRC divided by basal OCR; n = 5–6, *p < 0.05 unpaired Student’s t test. BMDC were pulsed or not with OVA323-339 peptide (1 μg/ ml) overnight and incubated with ADAM17 inhibitor 2155-17 (50 μM), ADAM10 inhibitor or meprin (20 μM), collected and cocultured for 4 days with CFSE-labeled splenic CD4 + naïve OTII cells (1:5). (C) Bar graph shows the frequency of Ag-specific proliferation and (D) IL13 released in the culture supernatant. (E) Representative flow cytometry analysis and (F) frequencies of CD4 + GATA3 + OTII cells after 4 days of coculture. Results show mean + / − S.D. and represent two or more independent experiments, n = 5–8 per group, *p < 0.05, ns not significant, one-way ANOVA with Tukey’s multiple comparison tests.

The adoptive transfer of sensitized DC treated with ADAM17 inhibitor 2155-17 has an impaired ability to induce HDM-mediated airway inflammation. (A) Experimental design for adoptive transfer. BMDC from donor 990 WT mice were pulsed with HDM (100 μg/ ml) in the presence or absence of 2155-17 (50 μM). About 75,000 viable CD11c + cells were transferred intranasally into recipient naïve WT mice. Daily intranasal HDM challenges (50 μg) were administered to all recipient mice on days 9, 11, 13, 15, and endpoints were analyzed on day16. (B) Number of total BAL cells and inflammatory cell types (CD11b + Siglec-F + eosinophils and CD11b + Ly-6G + neutrophils), (C) BAL CCL24, and (D) HDM-specific serum antibody levels from HDM-challenged mice that had been sensitized with CD11c + BMDC treated with 2155-17 and pulsed with either HDM or PBS. (E) Cytokine production (IL4, IL5, and IL13) by lung draining lymph nodes (DLn) cells from asthmatic mice restimulated ex vivo with HDM (100 μg/ ml) for 4 days. (F) Representative H&E and PAS staining of lung sections from WT recipient mice receiving HDM-sensitized BMDC treated with or without 2155 17 from donor mice. Scale bars, 100 μm for × 100 and 50 μm for × 400 images. Results are expressed as mean + / − SEM and represent one of the two adoptive transfer experiments; n = 4 to 5 mice per group; *p < 0.05, one-way ANOVA with Tukey’s multiple comparison test.
Inhibition of ADAM17 in vivo following antigen priming suppress the manifestations of HDM-induced airway disease and type 2 immune response
We employed a therapeutic model of HDM-induced experimental asthma to test whether local application of ADAM17 inhibitor 2155-17 abrogates airway disease in WT mice as previously described24. A cohort of mice received 2155-17 (200 μg per mouse; i.n.) treatment with HDM sensitization succeeding T cell priming had already occurred (Fig. 7A). HDM-sensitized WT mice that received 2155-17 concurrent with intranasal HDM challenges, but after allergic sensitization had already eventuated, demonstrated a significant reduction in inflammatory cells and eosinophils in the BAL and lungs (Fig. 7B). Analysis of peripheral blood showed marked decreases in HDM- specific IgG1, and IgE antibody levels in 2155-17 treated allergic mice (Fig. 7C). In parallel, analysis of lung cytokines showed a significant reduction in Th2 cytokines, IL5, and IL13 (Fig. 7D). Lung histopathology showed a reduction in the peri-bronchial inflammatory cell infiltrates scattered around large conducting airways and mucous cell metaplasia in 2155-17 treated allergic mice (Fig. 7E). Therefore, these results demonstrate that inhaled pharmacological ADAM17 inhibitor only during the challenge phase can similarly attenuate the manifestations of HDM-induced Th2/eosinophilic airway inflammation, thereby establishing ADAM17 as a potential therapeutic target in asthma development.

Inhaled ADAM17 inhibitor, 2155-17, prevented HDM-induced asthma exacerbation. (A) Experimental layout of ADAM17 inhibitor (2155-17) treatment in vivo. WT mice were sensitized and challenged with HDM to induce experimental asthma and were concurrently received intranasal ADAM17 inhibitor 2155-17 (200 μg per mouse) or PBS (Veh) on days 8, 10, 12, and 14 before endpoint assessment on day 16. (B) Enumeration of total inflammatory cells, CD11b + Siglec-F + eosinophils, and CD11b + Ly-6G + neutrophils in BAL (left) and lung (right). The bar graph represents (C) serum levels of HDM-specific IgG1 and IgE, and the scatter plot shows (D) lung cytokine levels from local application of 2155-17 in asthmatic mice. Each circle represents individual mice. (E) Representative histologic lung sections of 2155-17- and PBS treated asthmatic lung stained with PAS. Scale bars, 100 μm for × 100 and 50 μm for × 400 images. Results are expressed as mean + / − SEM and represent one of the two independent experiments; n = 4 to 7 mice per group; *p < 0.05 unpaired Student’s t-test.
Discussion
A principal finding of this study is the demonstration by both cDC-specific genetic KO (ΔDC mice) and chemical biology approaches that inhibition of ADAM17 can protect against disease-relevant type2/ eosinophilic airway inflammation. First, we show that cDC-specific ablation of ADAM17 suppresses type2/ eosinophilic polarized HDM allergic responses, which is the pathological correlate of asthma. Second, HDM induces metabolic reprogramming in DC coupled to ADAM17 activity, which affects the capacity to present antigen to CD4 + T cells and promotes type2 inflammatory response. Third, ADAM17 selective small-molecule inhibitor (2155-17) that acts via secondary site binding (non-zinc binding inhibition) strongly reduced allergen-specific OTII cell proliferation and production of type2 cytokines. In line with this, adoptive transfer of 2155-17-treated DC to naïve mice reverses the defect and attenuates type2/ eosinophilic polarized HDM allergic immune responses compared to vehicle-treated control (data not shown). We also showed initiation of 2155-17 treatment after T cell priming had already occurred, similarly reduced HDM-induced airway inflammation and type2 cell-mediated immune responses. The convergent results from two independent approaches but common histological and pathological endpoints demonstrate that ADAM17 plays a distinct regulatory role in lung DC and can be probed by inhibitors, thereby providing the foundation for a novel immune-modulating therapeutic strategy in an area of unmet medical need for asthma research.
cDC are directly involved as pivotal players controlling type2 immune responses in allergic asthma24,33. Recognition of allergens, activation, and migration of antigen-presenting airway DC to the draining lymph nodes (DLn) is indispensable for initiating and propagating T cell-mediated immunity in the airways. cDC subsets (CD11b + cDC2 and CD103 + cDC1) in the airways that specifically express the transcription factor Zbtb4627,29 are central for the induction of type 2- mediated eosinophilic airway inflammation to inhaled HDM . In addition, CD11b + monocyte-derived DC (moDC) primarily mediates pro-inflammatory cytokine productions, however, could also induce type2 immunity to inhaled HDM in higher doses39,40,41. Recent reports from our lab identified specific changes in the intrinsic function of DC and their progenitors, which are coupled to ADAM17 enzyme activity and are required for the proliferation and differentiation of DC- restricted progenitors25. Here we show that mice bearing floxed ADAM17 with a Zbtb46 Cre deleter strain (∆DC mice) manifested reduced lung function (AHR) and type2/ eosinophilic polarized HDM allergic responses. We demonstrate that non-allergic WT mice sensitized with HDM-pulsed DC isolated from ∆DC mice displayed attenuated airway inflammation associated with impaired type2 immune responses. DC maturation is coupled with metabolic reprogramming that fine-tunes MHCII-restricted antigen presentation and activates CD4 + cells to produce effector type 2 cytokines30,42. We show that a reduced state of metabolic activity in DC isolated from ∆DC mice impacted DC immune-priming function and suppresses allergen- specific T cells, suggesting the causal role of DC-specific ADAM17 activity for mounting eosinophilic asthma. Furthermore, we found that neither allergen uptake nor subsequent migration to DLn was impaired in CD11b + DC from ∆DC mice, which are the specialized DC subset for antigen presentation and type2 immune responses43. Additionally, we demonstrated a decrease of IL-13 and IL-4 secretion by BMDC from ΔDC mice, suggesting this defect in DC is limited to Th2 cytokine production44. We propose ADAM17 as a potential target in airway inflammation and a key determinant of HDM-induced Th2 cytokine secretion from DCs. However, further studies are warranted to understand the mechanism underlying ADAM-17-mediated cytokine secretion and regulation in DCs. IL-4/IL-13/IL-4Rα axis is a therapeutic target of Dupilumab, a fully human monoclonal antibody, which is approved for the treatment of asthma, atopic dermatitis, and chronic sinusitis with nasal polyposis45. This potentially means that ADAM17 inhibition can target the same indication with underlying type 2 signatures. Our study further extends the current understanding of ADAM17 biology and demonstrates that ADAM17 plays a distinct regulatory role in airway DC and controls metabolic adaptations of DC maturation/ activation upon HDM challenge, thereby accelerating type2/ eosinophilic allergic responses in asthma.
Earlier studies have linked a strong correlation between ADAM8, ADAM33, and ADAM10 in allergen-induced airway inflammation and asthma46,47,48,49,50. ADAM17 is a membrane-anchored metalloproteinase enzyme associated with the cleavage of more than eighty different substrates, thereby altering their biology and impacting immune and inflammatory responses in numerous lung diseases, including cigarette smoke-induced emphysema, COPD, cystic fibrosis, and LPS-induced acute lung injury16,17,51.
Because of the broad substrate cleavage potential, prior studies have focused on intrinsic ADAM17 sheddase activity to adhesion molecules, growth factors, cytokines, or their cognate membrane-bound receptors coupled with pro-inflammatory events14,52. In patients with allergic asthma, increased levels of ADAM10 and soluble CD23 (sCD23) are associated with type2 immune responses and the severity of the disease53. Interestingly, it has been shown that ablation of ADAM10 abrogates the priming potential of CD11c + antigen-presenting lung cells in a Notch1-dependent manner and is restored by Notch1 overexpression46. In agreement with this, we demonstrate that genetic ablation of ADAM17 in DC or blockade of ADAM17 activity by the non-zinc binding small molecule inhibitor 2155-17 significantly impacted HDM-induced mitochondrial respiratory activity in DC and influenced maturation/activation, which was absent by ADAM10 or meprin blockade. Mechanistically, we show that these upstream events are coupled to ADAM17 activity and regulate the immunogenic capacity of DC to prime allergen-specific T cell stimulation and the development of airway inflammation. Blockade of Notch2 impairs the migration of DC to DLn by modulating the expression of the chemokine receptor CCR7 and via CD44-like molecule lymphatic vessel endothelial hyaluronic acid receptor 1 (LYVE1), which are the substrates for both ADAM10 and ADAM1754. Our data reveal that cDC-specific ADAM17 deletion prevented the type2 immune responses to inhaled HDM were not due to differences in the capacity of allergy-producing CD11b + DC subsets to uptake and transport allergen-derived antigens to DLn but to their functional potential for immune-priming and immune-polarization. More than 50% of adult asthmatic patients clinically manifest eosinophilic/ type 2 airway inflammation than neutrophilic/ non-type 2 airway inflammation, which is prevalent only in 15% of cases1,55. Current pharmaceutical interventions to treat asthma are almost exclusively designed to alleviate symptoms. However, airway inflammation plays a seminal role in asthma development and progression. Although inhaled corticosteroids have been the foundation of asthma therapy, few treatment options exist for type2-high patients with severe asthma exacerbation5,8. Recent studies of type2 cytokine inhibition by multiple monoclonal antibodies and type2 biologic agents returned negative results, suggesting the heterogeneity and complexity of immunoregulatory pathways in asthma7,12. We propose that ADAM17 is an upstream target and can be probed by inhibitors for therapeutic prevention of type2-high allergic asthma. Here, we demonstrate that a selective small-molecule ADAM17 inhibitor that acts via a novel mechanism (secondary site binding, non-zinc-binding) renders an advantage to avoid off- target inhibition of a multitude of other zinc-dependent metalloproteinases, which plagued preceding generations of inhibitors. Studies from broad-spectrum metzincin inhibitors (e.g., BMS-561392) failed due to the lack of selectivity for the zinc-binding mechanism36. Another advantage of non-zinc-binding inhibition is substrate selectivity, which is unachievable with zinc- binding inhibitors. Therefore, it ensures their unique selectivity and safety profile and suggests lead-likeness and in vivo potential to prevent airway inflammation. To avoid systemic effects, we administered site-directed delivery of ADAM17 inhibitor 2155-17 directly to the lungs via inhalation after T cell priming had already occurred. We assessed whether pharmacological inhibition represents an effective therapeutic approach in vivo to mimic the protection seen with the targeted gene KO to HDM-induced allergic asthma. Our results demonstrate that the ADAM17 lead inhibitor 2155-17 similarly protected HDM-induced eosinophilic/ type 2 airway inflammation, confirming the KO mouse results. Thus, our findings lay out a foundation for future development of similar small-molecule inhibitors that are in vivo modulators of biological function.
Conclusion
Taken in its entirety, our report, to the best of our knowledge, is the first to provide critical proof-of-concept for ADAM17 contribution in DC-mediated allergic asthma that converged from two independent experimental approaches of targeted genetic KO and highly selective ADAM17-specific inhibitor results. Furthermore, we have identified a selective small-molecule ADAM17 inhibitor that acts via a conceptually novel mechanism of secondary exosite binding inhibition, thereby rendering immune-priming function(s) of DC and diminishing type 2 allergic asthma. This identifies ADAM17 as one of the stream mediators in airway inflammation and a potential therapeutic target that can serve as a new treatment in conjunction with other therapies for patients with type 2- high allergic asthma.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
Abbreviations
- ADAM17:
-
A disintegrin and metalloproteinase domain 17
- DC:
-
Dendritic cells
- HDM:
-
House dust mite
- AHR:
-
Airway hyperresponsiveness
- WT:
-
Wild-type
References
Masoli, M., Fabian, D., Holt, S. & Beasley, R. The global burden of asthma: Executive summary of the GINA dissemination committee report. Allergy Eur. J. Allergy Clin. Immunol. 59, 469–478 (2004).
Reibman, J. et al. Clinical and economic burden of severe asthma among US patients treated with biologic therapies. Ann. Allergy Asthma Immunol. 127 (3), 318–325 (2021).
Wenzel, S. E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 18, 716–725 (2012).
Pavlidis, S., et al. “T2-high” in severe asthma related to blood eosinophil, exhaled nitric oxide and serum periostin. Eur. Respir. J. 53 (1), (2019).
Brusselle, G. G. & Koppelman, G. H. Biologic therapies for severe asthma. N. Engl. J. Med. 386 (2), 157–171 (2022).
Reddel, H. K. et al. Global Initiative for asthma strategy 2021 executive summary and rationale for key changes. Am. J. Respir. Crit. Care Med. 205 (1), 17–35 (2022).
Yılmaz, I. & Türk, M. Biologic targeted therapies for the phenotypes of type-2 high severe asthma. J. Allergy Clin. Immunol. Pract. 6, 1091–1092 (2018).
Crossingham, I. et al. Combination fixed-dose β agonist and steroid inhaler as required for adults or children with mild asthma: A cochrane systematic review. BMJ Evid. Based Med. 27, 178–184 (2022).
Ortega, V. E. et al. Pharmacogenetic studies of long-acting beta agonist and inhaled corticosteroid responsiveness in randomised controlled trials of individuals of African descent with asthma. Lancet Child Adolesc. Heal. 5 (12), 862–872 (2021).
McDowell, P. J. et al. The inflammatory profile of exacerbations in patients with severe refractory eosinophilic asthma receiving mepolizumab (the MEX study): a prospective observational study. Lancet Respir. Med. 9 (10), 1174–1184 (2021).
Leckie, M. J. et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 356 (9248), 2144–2148 (2000).
Wenzel, S. E. Severe adult asthmas: Integrating clinical features, biology, and therapeutics to improve outcomes. Am. J. Respir. Crit. Care Med. 203, 809–821 (2021).
Schumacher, N. & Rose-John, S. ADAM17 orchestrates Interleukin-6, TNFα and EGF-R signaling in inflammation and cancer. Biochim. Biophys. Acta Mol. Cell Res. 1869 (1), 119141 (2022).
Moss, M. L. & Minond, D. Recent advances in ADAM17 research: A promising target for cancer and inflammation. Mediat. Inflamm. 2017, 9673537 (2017).
Tang, B. et al. Substrate-selective protein ectodomain shedding by ADAM17 and iRhom2 depends on their juxtamembrane and transmembrane domains. FASEB J. 34 (4), 4956–4969 (2020).
Dreymueller, D. et al. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol. Med. 4 (5), 412–423 (2012).
Park, J. A. et al. Human neutrophil elastase-mediated goblet cell metaplasia is attenuated in TACE-deficient mice. Am. J. Physiol. Lung Cell Mol. Physiol. 304 (10), L701–L707 (2013).
Van Barneveld, R. J., Dunshea, F. R., Beddhu, S., Filipowicz. R., Chen X. X. W. X. X., Neilson, J. L., et al. Bone health in gynecologic cancers-does FOSAVANCE Help? Am. J. Clin. Nutr. (2019).
Zhao, J. et al. Pulmonary hypoplasia in mice lacking tumor necrosis factor-α converting enzyme indicates an indispensable role for cell surface protein shedding during embryonic lung branching morphogenesis. Dev. Biol. 232 (1), 204–218 (2001).
le Cras, T. D. et al. Epithelial EGF receptor signaling mediates airway hyperreactivity and remodeling in a mouse model of chronic asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 300 (3), L414–L421 (2011).
Amishima, M. et al. Expression of epidermal growth factor and epidermal growth factor receptor immunoreactivity in the asthmatic human airway. Am. J. Respir. Crit. Care Med. 157 (6), 1907–1912 (1998).
Hammad, H. et al. Inflammatory dendritic cells - not basophils - are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J. Exp. Med. 207 (10), 2097–2111 (2010).
Mishra, A. et al. Low-density lipoprotein receptor-related protein 1 attenuates house dust mite-induced eosinophilic airway inflammation by suppressing dendritic cell-mediated adaptive immune responses. J. Allergy Clin. Immunol. 142, 1066–1079 (2018).
Mishra, A. et al. Dendritic cells induce Th2-mediated airway inflammatory responses to house dust mite via DNA-dependent protein kinase. Nat. Commun. 6, 6224 (2015).
Jaiswal, A. K. et al. Dendritic cell-restricted progenitors contribute to obesity-associated airway inflammation via Adam17-p38 MAPK-dependent pathway. Front. Immunol. 11, 363 (2020).
Sichien, D., Lambrecht, B. N., Guilliams, M. & Scott, C. L. Development of conventional dendritic cells: From common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol. 10, 831–844 (2017).
Satpathy, A. T. et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 209 (6), 1135–1152 (2012).
Zhang, J. et al. Bone marrow dendritic cells regulate hematopoietic stem/progenitor cell trafficking. J. Clin. Invest. 129 (7), 2920–2931 (2019).
Meredith, M. M. et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med. 209 (6), 1153–1165 (2012).
Jaiswal, A. K. et al. Irg1/itaconate metabolic pathway is a crucial determinant of dendritic cells immune-priming function and contributes to resolute allergen-induced airway inflammation. Mucosal Immunol. 15, 301–313 (2022).
Knapinska, A. M. et al. SAR studies of exosite-binding substrate-selective inhibitors of A disintegrin and metalloprotease 17 (ADAM17) and application as selective in vitro probes. J. Med. Chem. 58 (15), 5808–5824 (2015).
Lauer-Fields, J. L. et al. High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triple-helical FRET substrate. Bioorg. Med. Chem. 17 (3), 990–1005 (2009).
Plantinga, M. et al. Conventional and monocyte-derived CD11b+ dendritic cells initiate and maintain T Helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38 (2), 322–335 (2013).
Jaiswal, A. K. et al. Pyruvate kinase M2 in lung APCs regulates Alternaria-induced airway inflammation. Immunobiology 225 (4), 151956 (2020).
Naus, S. et al. The metalloprotease-disintegrin ADAM8 is essential for the development of experimental asthma. Am. J. Respir. Crit. Care Med. 181 (12), 1318–1328 (2010).
Georgiadis, D. & Yiotakis, A. Specific targeting of metzincin family members with small-molecule inhibitors: Progress toward a multifarious challenge. Bioorg. Med. Chem. 16, 8781–8794 (2008).
Fingleton, B. Matrix metalloproteinases as valid clinical target. Curr. Pharm. Des. 13 (3), 333–346 (2006).
Minond, D. et al. Discovery of novel inhibitors of a disintegrin and metalloprotease 17 (ADAM17) using glycosylated and non-glycosylated substrates. J. Biol. Chem. 287 (43), 36473–36487 (2012).
Vander Lugt, B. et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat. Immunol. 15 (2), 161–167 (2014).
Mesnil, C. et al. Resident CD11b+Ly6C- lung dendritic cells are responsible for allergic airway sensitization to house dust mite in mice. PLoS ONE 7 (12), e53242 (2012).
Bosteels, C. et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity 52 (6), 1039–1056 (2020).
Garcia Cruz, D. et al. Lymphocyte activation gene-3 regulates dendritic cell metabolic programing and T cell priming function. J. Immunol. 207 (9), 2374–2384 (2021).
Madoux, F. et al. Discovery of an enzyme and substrate selective inhibitor of ADAM10 using an exosite-binding glycosylated substrate. Sci. Rep. 6 (1), 11 (2016).
Lownik, J. C., Martin, R. & Conrad, D. A disintegrin and metalloproteinase 17 is required for IL-33 induced ILC2 expansion and cytokine production. J. Immunol. 200 (1), 104–107 (2018).
Le Floch, A. et al. Dual blockade of IL-4 and IL-13 with dupilumab, an IL-4Rα antibody, is required to broadly inhibit type 2 inflammation. Allergy Eur. J. Allergy Clin. Immunol. 75 (5), 1188–1208 (2020).
Damle, S. R. et al. ADAM10 and Notch1 on murine dendritic cells control the development of type 2 immunity and IgE production. Allergy Eur. J. Allergy Clin. Immunol. 73 (1), 125–136 (2018).
Shapiro, S. D. & Owen, C. A. ADAM-33 surfaces as an asthma gene. N Engl J Med. 347 (12), 936–938 (2002).
Post, S. et al. ADAM10 mediates the house dust mite-induced release of chemokine ligand CCL20 by airway epithelium. Allergy Eur. J. Allergy Clin. Immunol. 70 (12), 1545–1552 (2015).
Chen, J. et al. A novel peptide ADAM8 inhibitor attenuates bronchial hyperresponsiveness and Th2 cytokine mediated inflammation of murine asthmatic models. Sci. Rep. 6, 30451 (2016).
Oreo, K. M. et al. Sputum ADAM8 expression is increased in severe asthma and COPD. Clin. Exp. Allergy 44 (3), 342–352 (2014).
Saad, M. I. et al. ADAM17 deficiency protects against pulmonary emphysema. Am. J. Respir. Cell. Mol. Biol. 64 (2), 183–195 (2021).
Scheller, J., Chalaris, A., Garbers, C. & Rose-John, S. ADAM17: A molecular switch to control inflammation and tissue regeneration. Trends Immunol. 32, 380–387 (2011).
Cooley, L. F. et al. Increased B cell ADAM10 in allergic patients and Th2 prone mice. PLoS ONE 10 (5), e0124331 (2015).
Nishida-Fukuda, H. et al. Ectodomain shedding of lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1) is induced by vascular endothelial growth factor A (VEGF-A). J. Biol. Chem. 291 (20), 104900–110500 (2016).
Demarche, S. et al. Detailed analysis of sputum and systemic inflammation in asthma phenotypes: Are paucigranulocytic asthmatics really non-inflammatory?. BMC Pulm Med. 16 (1), 46 (2016).
Acknowledgements
We thank Dr. Maninder Sandey at the Department of Pathobiology, College of Veterinary Medicine, AU, AL, USA for support and advice. We also thank for the funding support under Award Number R03AI168864-01 to A.M from National Institutes of Health, USA.
Funding
Open access funding provided by Siksha 'O' Anusandhan (Deemed To Be University). The funding support was under Award Number R03AI153794-01A1 to A.M. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health to A.M (2021–23). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
A.K.J, D.M and A.M. designed, and analysed the data. A.K.J, and D.M. performed the experiments and A.M., and D.M. drafted the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jaiswal, A.K., Minond, D. & Mishra, A. Targeting ADAM17 to dampen dendritic cell-mediated type 2 immune responses and airway inflammation associated with allergic asthma. Sci Rep 15, 31253 (2025). https://doi.org/10.1038/s41598-025-14569-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14569-w
