
Abstract
[18F]-Fluoroestradiol, which is recently approved by the FDA, is a well-recognized estrogen receptor radiopharmaceutical in nuclear medicine to investigate both primary and metastatic breast cancers. This study introduces an innovative and effective method for synthesizing the DOTA-coupled estradiol derivative and evaluating its preclinical potential. 68Ga/177Lu-labeled estradiol derivative was evaluated in vitro for its stability in human plasma, and binding capacity was determined using the ER-positive MCF-7 breast cancer cell line. In vivo, biodistribution and tumor targeting were conducted in ER-positive MCF-7 tumor-bearing nude mice. DOTA conjugated estradiol was prepared conveniently using solid-phase synthesis, and its radiolabeling with both 68Ga and 177Lu resulted in the formation of one major product, with high labeling efficiency (≥ 90%). Also, a high stability of the radiotracer was found in human plasma. The radiolabeled estradiol derivative exhibited a nanomolar affinity (< 20 nM) specific to the MCF-7 cell line. In the MCF-7 tumor xenograft model, the radioestradiol derivative displayed fast clearance from the blood and excretion mainly by the renal system. A rapid accumulation of 3.79% IA/g was observed in the MCF-7 tumors at 1 h p.i., whereas a low to moderate retention of radioactivity was seen in the normal organs, including the heart, lungs, liver, stomach, spleen, intestines, and kidneys (< 5% ID/g). The receptor specificity of the radioestradiol was confirmed by the receptor-blocking assay. The rapid and efficient targeting of tumors, along with the favorable pharmacokinetics, emphasizes the potential of radioestradiol for targeting tumors. Additionally, PET imaging demonstrated good visualization of MCF-7 tumors in nude mice. Our results indicate that the 68Ga/177Lu-labeled estradiol derivative could serve as a promising radiopharmaceutical option for the effective targeting of MCF-7 tumors.
Similar content being viewed by others


Introduction
The development of new and innovative radiopharmaceuticals has made significant strides in the last decade, particularly for personalized nuclear medicine, using PET (positron-emission tomography) and SPECT (single-photon emission computed tomography). As a result, there are numerous potent radiopharmaceuticals used in nuclear oncology for diagnostic imaging and targeted treatment of cancer, and that number is still growing. A recent and notable trend of growing the number of receptor-targeting radiopharmaceuticals is because receptor binding agents hold the ability to highly and specifically accumulate in the target receptor site, enabling highly sensitive and precise detection of receptor-positive tumors using PET. Many of these receptor-targeting agents are small-sized bioactive molecules that typically exhibit rapid pharmacokinetics, aligning well with radionuclides that have suitable half-lives, to maximize their clinical utility for diagnostic imaging and targeted radionuclide therapy1,2. One example of a bioactive molecule that binds to receptors is [18F]-Fluoroestradiol, which has a high affinity and specificity for estrogen-receptor-positive breast cancer.
In patients with breast cancer, estrogen-receptor (ER) expression is crucial for determining risk and forecasting treatment response. Estrogen-receptor testing of lesions is advised for any primary and newly metastatic breast cancer by the American Society of Clinical Oncology (ASCO) and the US National Comprehensive Cancer Network (NCCN). Nearly 75% of all breast cancers in women, and 99% in men, are categorized as ER-positive, due to an overexpression of the ER3,4,5,6. As a result, determining the ER content of human breast cancers has proven to be useful in determining the best course of action for treating metastatic breast carcinomas. Hormonal treatment is generally ineffective for cancers that don’t have estrogen receptors, but it is more effective for tumors with estrogen receptors7,8. It is well established that through a complicated signaling cascade, estrogen promotes cell proliferation and prevents apoptosis, leading to transcriptional changes that may include the modification of tumor suppressor activity. The ER offers a potential mechanism for the selective accumulation of estrogens within estrogen receptor-positive tumors since it is a protein that binds estrogens with high affinity7,8,9. It is widely recognized that early and precise diagnosis of cancer, along with the assessment of estrogen receptor status and metastatic condition, can lead to better patient outcomes with the appropriate treatment10,11,12.
Hormonal therapy may be effective for ER-positive tumors at the diagnosis stage, but chemotherapy or surgery is necessary for ER-negative tumors. Immunohistochemistry is frequently used after biopsy to determine the ER’s state. However, the accuracy of this invasive approach for detecting tumor metastases is limited. Non-invasive molecular imaging modalities, such as PET, provide an effective means to visualize the entire tumor heterogeneity by imaging ER-positive lesions, thereby addressing these limitations13. Given that ER is overexpressed in 75% of breast carcinomas, it represents a promising molecular target for both cancer diagnosis and treatment. Thus, several PET and SPECT-based agents radiolabeled with, for example, 18F9,14,15, 99mTc10,16,17, or 131I18 have been prepared and evaluated for targeting ER-positive tumors. Presently, the most effective [18F]-labeled estrogen derivative that has undergone clinical evaluation is [18F]-Fluoroestradiol ([18F]-FES), a steroid-based PET radiotracer. This PET radiotracer has demonstrated encouraging outcomes in the imaging of estrogen-receptive tumors and in forecasting the sensitivity of breast tumors to antiestrogen therapies, including tamoxifen19,20. The US FDA has recently approved [18F]-FES to be used as a radioactive diagnostic agent for identifying ER-positive primary and metastatic breast cancer lesions21,22.
This work was initiated to develop an estradiol-based biomolecule linked to a DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) chelator that enables radiolabeling with both diagnostic and therapeutic radionuclides. This paper discusses the synthesis, radiolabeling, and both in vitro and in vivo assessment of a newly developed DOTA-coupled estradiol derivative.
Results
Synthesis of estradiol derivative
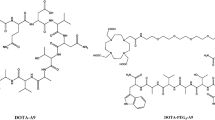
The estradiol derivative was prepared successfully by first reacting estrone with tert-butyl bromoacetate to give tert-butyl protected estradiol acetate, which, upon treatment with TFA/ DCM, provided acetic acid estradiol. This intermediate precursor was further reacted with the alpha-amino group of Lys(ivDde)-OH to provide lysine acetic acid estradiol, whereas the epsilon-amino group of the lysine acetic acid estradiol was reacted with DOTA to yield the target DOTA-estradiol compound. The overall synthetic yield was 47%. The structural identity of the DOTA-estradiol was confirmed by mass spectrometry and found to be consistent with the proposed chemical structure.
Radiolabeling with 68Ga and 177Lu
DOTA-coupled estradiol was radiolabeled efficiently with 68Ga simply by dissolving the estradiol in sodium acetate buffer, followed by the addition of [68Ga]GaCl3 and heating the reaction mixture to accelerate the radiolabeling kinetics. Radiolabeling with [177Lu]LuCl3 was performed nearly in a similar manner, except using preferably ammonium acetate buffer and the addition of gentisic acid to the reaction mixture to minimize the possible radiolysis. These facile procedures provide reproducible and efficient radiolabeling (≥ 90%) of estradiol via DOTA chelator with both 68Ga and 177Lu radionuclides, with molar radioactivity of greater than 300 Ci/mmol. It is evident from the radio-HPLC analyses that the [68Ga]- and [177Lu]-labeled estradiol formed mainly one radioactive species, with retention times of 15.28 and 15.35 min, respectively. Minor peaks of free 68Ga and free 177Lu were eluted at around 3–4 min. The [68Ga/177Lu]-labeled estradiol derivatives have shown stability for at least 4 h and 24 h, respectively, at room temperature as revealed by radio-HPLC analysis. Additionally, the identification of the [68Ga]-DOTA-estradiol complex was confirmed by co-injecting the nonradioactive natGa-DOTA-estradiol, as it displayed a nearly similar retention time UV profile to that of [68Ga]-DOTA-estradiol under the same HPLC conditions. Representative HPLC chromatograms are presented in Fig. 1.

Representative radio-HPLC elution profile of the [68Ga]-DOTA-estradiol at 45 min post-labeling (upper); and [177Lu]-DOTA-estradiol at 60 min post-labeling (middle), and the reference natGa-DOTA-estradiol for the identity confirmation at UV 220 nm (lower).
In vitro metabolic stability in human plasma
The [68Ga/177Lu]-labeled estradiol derivatives were incubated with human plasma at 37 °C for up to 4 h to assess the extent of proteolytic breakdown. The radio-HPLC analysis revealed that more than 80% of the radioactivity was still bound to the [177Lu]-estradiol after 4 h of incubation (Fig. 2). The [68Ga]-estradiol exhibited a nearly similar stability profile with human plasma at 2.5 h incubation (Fig. 3). The high metabolic stability of the [68Ga/177Lu]-labeled estradiol was demonstrated by the modest release of free 177Lu or 68Ga (up to 20%), with minimal enzymatic breakdown by plasma proteases.

Representative radio-HPLC chromatograms of the [177Lu]-DOTA-estradiol after 4 h incubation with human plasma (upper); radio-HPLC analysis of a mouse urine sample collected at 4 h after injecting the [177Lu]-DOTA-estradiol (lower).

Representative radio-HPLC chromatograms of [68Ga]-DOTA-estradiol after 2.5 h incubation with human plasma (upper); radio-HPLC analysis of a mouse urine sample collected at 2.5 h post-injection of [68Ga]-DOTA-estradiol (lower).
Lipophilicity determination
Log P values of the [68Ga/177Lu]-labeled estradiol derivatives were determined using the octanol-water distribution method (Table 1). The ratio of octanol layer counts to aqueous layer counts was expressed as the distribution coefficient. The obtained Log P values of − 0.70 ± 0.09 for [68Ga]-estradiol and − 0.92 ± 0.11 for [177Lu]-estradiol, suggesting that both of these estradiol derivatives are rather hydrophilic complexes. When compared with the reported Log P value of the reference [18F]-FES, (3.3‒5.0)10,23, the [68Ga/177Lu]-labeled estradiol derivatives evaluated in this study were found to be more hydrophilic (-0.70 to -0.92).
In vitro tumor cell binding and cell internalization assays
Receptor binding and cell internalization assays were performed with MCF-7 breast cancer cells, and MDA-MB-231 was used as a negative control. The cell binding data (Table 1) showed that the [68Ga]-estradiol exhibited the binding affinity (Kd) value of 17.68 ± 3.74 nM to the ER-positive MCF-7 cell line examined in this study (Fig. 4). A slightly better binding affinity pattern was observed for [177Lu]-estradiol over [68Ga]-estradiol, with a binding affinity value of 14.38 ± 3.30 nM for the MCF7 cell line. These bindings were significantly blocked or reduced in the presence of a 200-fold excess of unlabeled estradiol or with the MDA-MB-231 cell line, suggesting that the binding is specific. Overall, the binding of the [68Ga/177Lu]-labeled estradiol derivatives to breast cancer cells was determined to be saturable and specific to receptors.

In vitro saturation cell binding calculation of [68Ga/177Lu]-DOTA-estradiol derivatives with the ER-positive MCF-7 cancer cell line. [68Ga]-DOTA-estradiol showed a binding affinity of 17.68 nM (left), whereas its [177Lu]-DOTA-estradiol counterpart (right) displayed a somewhat better cell binding affinity, with the Kd value of 14.38 nM (n = 3).
Cellular internalization
In vitro internalization studies were performed to determine the rate of internalization of the [177Lu]-estradiol into MCF-7 human breast cancer cells (Table 1). Following incubation, the cells were washed with sodium acetate buffer (pH 5.0) to remove the surface-bound radioactivity. The results of the internalization experiments showed that 20 ± 4.29% of the total cell-bound activity of the [177Lu]-estradiol was internalized into the MCF-7 cells.
Biodistribution studies
The in vivo biodistribution studies of the [68Ga/177Lu]-labeled estradiol derivatives were carried out in female Balb/c mice, at two different time points, to observe the physiological uptake in the normal organs/tissues and to explore the rate and route of the excretion of the radiolabeled estradiol. The biodistribution results are presented in Fig. 5. For comparison purposes, the biodistribution of the [18F]-FES is also performed and shown in Fig. 5. For the [68Ga]-estradiol, rapid clearance from the blood was observed both at 45 min and 2.5 h p.i., with less than 0.5% IA/g of radioactivity retained in the blood after 2.5 h p.i. Moderate uptake and retention were found in the liver, with the values ranging from 2.90 ± 0.58% IA/g at 45 min to 0.89 ± 0.20% IA/g at 2.5 h p.i. A high amount of radioactivity was seen in the intestines, with the contents (up to 4.03% IA/g), both at 45 min and 2.5 h p.i. Uptake and retention by the kidneys were low at both time points (up to 2.08% IA/g). The tumor-targeting radiopharmaceutical that exhibited reasonably low kidney retention is preferred for diagnostic imaging and particularly for radionuclide therapy because of the potential kidney toxicity2. The uptake in the ER-rich tissue uterus was moderate (up to 1.93% IA/g). Low radiolabeled estradiol uptake in the bone (up to 0.80% IA/g) indicates the minimal breakdown of the [68Ga]-estradiol in vivo as well as the minor release of free 68Ga. These findings are as per the high in vitro metabolic stability obtained in the human plasma for the radiolabeled estradiol. The accumulation in the highly vascular lungs was also low (up to 0.91% IA/g), demonstrating a low trapping by the lungs. [68Ga]-estradiol was excreted mainly through the renal system. In vivo biodistribution of the [177Lu]-estradiol counterpart displayed practically the same biological and excretion profile as that of the [68Ga]-estradiol in Balb/c mice (Fig. 5), signifying the low influence of the radionuclides on the pharmacokinetic characteristics of the estradiol derivative (Fig. 5). Moreover, the biodistribution profile of [18F]-FES (Fig. 5) closely aligns with that of [68Ga/177Lu]-labeled estradiol regarding the uptake in the major organs, including the liver and intestines. However, the kidneys and uterus showed a somewhat greater level of radioactivity uptake.

Uptake of [68Ga/177Lu]-DOTA-estradiol derivatives in selected organs of female Balb/c mice. In vivo tissue biodistribution data were obtained at 45 min and 2.5 h for 68Ga-DOTA-estradiol (Top) and [18F]-FES (as a reference) (middle); and at 1 h and 4 h post-injection for [177Lu]-DOTA-estradiol (bottom). All uptake values are expressed as % IA/g. Data represent mean values ± SD from at least 4 determinations.
In vivo tumor targeting
Due to their suitable pharmacokinetic properties, the [68Ga/177Lu]-labeled estradiol derivatives were subjected to further investigation regarding their in vivo tumor-targeting capabilities in nude mice with ER-positive MCF-7 xenografts. In nude mice, the [68Ga]-estradiol also exhibited fast clearance from the blood, as only 0.30% IA/g of radioactivity was observed in the blood at 2.5 h p.i. (Fig. 6). A good and rapid uptake of 3.18 ± 0.76% IA/g was found in the MCF-7 tumors at 45 min p.i. The tumor showed good retention and reduced to 2.69 ± 0.57% IA/g at 2.5 h p.i. (with 16% washout from the tumors over 2.5 h). The uptake of the [68Ga]-estradiol in the tumors was found to be always higher than the uptake by the blood and muscle, resulting in reasonably good tumor-to-blood and tumor-to-muscle uptake ratios. A general pattern of better tumor-to-blood and tumor-to-muscle uptake ratios was noticed over time for [68Ga]-estradiol. The tumor-to-blood uptake ratio achieved was 7.22 at 45 min p.i., which increased to 9.61 at 2.5 h p.i. because of the rapid clearance from the blood, whereas the tumor-to-muscle uptake ratio was 15.90 at 45 min, which enhanced to 17.93 at 2.5 h p.i. The radioactivity uptake in the major body organs, such as the lungs, spleen, stomach, liver, intestines, and kidneys in nude mice was low to moderate (below 5% IA/g), and found to be somewhat comparable with normal Balb/c mice biodistribution (Fig. 5). The radioactivity found in the ER-positive uterus was moderate (up to 1.93% IA/g). The [68Ga]-estradiol displayed a low accumulation in the bone both at 45 min and 2.5 h p.i. (up to 0.27% IA/g), highlighting its high in vivo stability.

In vivo tumor targeting of [68Ga/177Lu]-DOTA-estradiol derivatives in female nude mice bearing ER-positive MCF-7 tumor xenografts. In vivo tumor targeting and tissue biodistribution data were obtained at 45 min and 2.5 h for [68Ga]-DOTA-estradiol (upper) and at 1 h and 4 h post-injection for [177Lu]-DOTA-estradiol (bottom). A blocking study was performed by co-injecting ~ 200 µg of unlabeled DOTA-estradiol with the [68Ga]-DOTA-estradiol at 2.5 h post-injection. All uptake values are expressed as % IA/g. Data represent mean values ± SD from at least 4 determinations.
The accumulation of [177Lu]-estradiol in ER-positive MCF-7 tumors was marginally superior to that of [68Ga]-estradiol. At 1 h p.i., the tumor uptake value was 3.79 ± 0.87% IA/g, which decreased to 2.55 ± 0.63% IA/g at 4 h p.i. (Fig. 6). The tumor-to-blood uptake ratio obtained was 9.48 at 1 h p.i., which enhanced to 9.81 at 4 h p.i. While the tumor-to-muscle uptake ratio was 34.45 and 36.43, respectively, at 1 h and 4 h p.i. The in vivo tumor targeting properties of the [177Lu]-estradiol were analogous to the [68Ga]-estradiol in MCF-7 xenograft models. Like the [68Ga]-estradiol, the main route of excretion of the [177Lu]-estradiol in the tumor-bearing mice was through the renal system, with the radioactivity excreted into the urine was up to 50% IA; whereas the clearance via the hepatobiliary system (liver + intestines) of was below 10% IA.
Receptor specificity study
We conducted a receptor-blocking analysis at 2.5 h p.i. to confirm the estradiol derivatives’ selectivity for their target estrogen receptors. A dose of ∼200 µg of nonradioactive estradiol premixed with the [68Ga]-estradiol was used for receptor blocking. It was observed that the blocking dose reduced the uptake in the tumors by approximately 62% (1.02 ± 0.19% IA/g blocked vs. 2.69 ± 0.62% IA/g unblocked, P = 0.011), highlighting the specificity of the [68Ga]-estradiol for the respective ER-positive breast cancer cells. Moreover, the uptake in the ER-rich uterus was reduced to approximately 46% (0.71 ± 0.15% IA/g blocked vs. 1.3 ± 0.22% IA/g unblocked, P = 0.088). Additionally, a variable effect of the pharmacological receptor-blocking dose was seen in the stomach, spleen, intestines, liver, and kidneys24 (Fig. 6).
In vivo metabolic stability
Athymic nude mice were used to study the in vivo metabolism as well as to determine the potential radio-metabolites in the urine. An aliquot of the urine sample was collected in a tube by manual voiding at the time of sacrifice and analyzed by radio-HPLC. Radio-HPLC analysis of the urine samples demonstrated that a major portion of the total radioactivity (> 70%) was still attached to the radioestradiol (Figs. 2 and 3). Some minor metabolite peaks detected at around 3–4 min represent the formation of free 68Ga or 177Lu release from the radiolabeled estradiol. This finding confirms that the radiolabeled estradiol derivatives exhibit resistance to rapid in vivo degradation and demonstrate a strong correlation with their high proteolytic stability observed in vitro.
PET imaging
The tumor imaging potential of the [68Ga]-estradiol was evaluated in female nude mice bearing subcutaneous ER-positive tumor xenografts at 45 min and 2.5 h p.i. after injecting 150 µCi (∼0.001 nmol, 100 µL) via the tail vein. Whole-body PET imaging studies revealed high uptake in the liver and intestines. Uptake in the MCF-7 tumor was relatively low when compared to the background activity. Additionally, high activity was seen in the urinary bladder (Fig. 7). The high urinary bladder activity of the [68Ga]-estradiol suggests its main pathway of excretion. The uterus was not seen, probably due to its overlapping with the intestine. Following the imaging study, the quantitative biodistribution was conducted to validate the results of the PET imaging. The PET imaging results were consistent with the quantitative biodistribution data summarized in Fig. 6.

Micro-PET camera image of a female nude mouse model with ER-positive MCF-7 tumor xenografts at 45 min post intravenous tail injection of 150 µCi of the [68Ga]-DOTA-estradiol. High abdominal activity can be seen in the image. The arrow indicates the tumor location.
Discussion
Breast cancer is a heterogeneous disease, and its heterogeneity can be reflected in its molecular characteristics and treatment results. Therefore, understanding the hormone receptor status is crucial, as it is present in approximately 75 to 80% of all breast cancer cases24,25. The stimulation of ERs by estrogen plays a critical role in the onset of ER-positive breast cancer. The status of ERs serves as a recognized prognostic and predictive biomarker, guiding the selection of treatment options involving hormone therapy agents that either inhibit ERs or reduce estrogen levels, thereby establishing ER as a valuable therapeutic target for managing ER-positive breast cancer. Over the last two decades, numerous radiolabeled compounds have been developed to specifically target ER in cases of ER-positive breast cancer; for instance, [18F]-FES has emerged as a leading radiotracer for the detection of ER-positive breast cancer19,21. Although [18F]-FES serves solely as a diagnostic radiotracer, the advancement of novel theranostic estradiol radiopharmaceuticals would greatly enhance the management of ER-expressing breast cancer. This study outlines an innovative and effective approach for synthesizing an estradiol derivative. We detail the synthesis, radiolabeling with both 68Ga and 177Lu, and the in vitro and in vivo assessments of the estradiol derivative to evaluate its capacity to target ER-positive MCF-7 breast cancer cell lines in preclinical studies.
The solid-phase synthesis method described here offers the significant benefit of producing estradiol efficiently, requiring fewer synthetic and purification steps, and offers a scalable and adaptable synthetic strategy that can be linked to various bifunctional ligands using cost-effective reagents. The resulting estradiol was purified through HPLC and analyzed via mass spectrometry. The DOTA-conjugated estradiol was successfully radiolabeled with both 68Ga and 177Lu radionuclides under mild acidic conditions (pH 5), resulting in reproducible and stable radiolabeled estradiol compounds suitable for in vitro and in vivo assessments.
High proteolytic stability is an important feature of the tumor-targeting biomolecules to deliver the maximal tumor-targeting effect to the tumor lesions. Indeed, our data suggest that the [68Ga/177Lu]-labeled estradiol exhibited high metabolic stability in human plasma in vitro, with low enzymatic degradation. A radiopharmaceutical’s lipophilicity is an important determinant of its possible nonspecific binding to non-targeted tissues and its excretion pathway. [68Ga/177Lu]-labeled estradiol displayed moderate hydrophilicity (Log P = ‒0.70 to ‒0.92), suggesting that these complexes are much more hydrophilic than the estradiol compounds reported in the literature (Log P = 3.3‒5.0)10,23.
In vitro cell binding was determined by employing the saturation binding assay, and the results were calculated by fitting the data with nonlinear regression using GraphPad Prism. [68Ga]-estradiol showed a binding affinity (Kd) of 17.68 nM, while its counterpart [177Lu]-estradiol exhibited a somewhat better binding profile, with the Kd value of 14.38 nM. [177Lu]-estradiol was further evaluated for cellular internalization and displayed that 20% of the total cell binding activity was internalized into MCF-7 breast cancer cells. It should be noted here that the binding affinities of the [68Ga/177Lu]-labeled estradiol derivatives to the human breast cancer cell lines were found to be about two times higher than those previously reported for the 99mTc-DTPA-estradiol, with a binding affinity value of 30 nM17. However, the binding affinity was found to be slightly lower than the one mentioned for the 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative (Kd = 11 nM)10. The results of the cell-binding and internalization into breast cancer cells show that the radiolabeled estradiol derivatives retained their maximum potency and held high affinities and specificities toward ER-positive breast carcinomas.
In vivo pharmacokinetics of the [68Ga/177Lu]-estradiol derivatives displayed rapid clearance from the blood. The kidneys showed low uptake and retention, while a somewhat high amount of radioactivity was found in the liver and intestines. Due to the known liver uptake of estradiol derivatives, we also performed the biodistribution of the reference [18F]-FES for comparison purposes. Nonetheless, our data suggest that the [68Ga/177Lu]-estradiol derivatives exhibited relatively lower liver and intestinal accumulation as compared to the [18F]-FES, possibly related to their higher hydrophilicity than the [18F]-FES.
[68Ga/177Lu]-labeled estradiol derivatives were evaluated for their in vivo tumor-targeting ability in nude mice with ER-positive MCF-7 xenografts. A good and fast uptake of 3.18 ± 0.76% IA/g was observed in the MCF-7 tumors at 45 min p.i. The tumor displayed good retention and decreased to 2.69 ± 0.57% IA/g at 2.5 h p.i. (with 16% washout from the tumors over 2.5 h). It was found that the uptake of the [68Ga]-estradiol was always higher than the uptake found in the blood and muscle, resulting in reasonably good tumor-to-blood and tumor-to-muscle uptake ratios. A general trend of better tumor-to-blood and tumor-to-muscle uptake ratios was observed over time for the [68Ga]-estradiol. The receptor specificity was confirmed by co-injecting an excess of non-radioactive estradiol. A slightly better tumor uptake was observed for the [177Lu]-estradiol, with 3.79 ± 0.87 and 2.55 ± 0.63, respectively, at 1 h and 4 h p.i. Both the [68Ga]- and [177Lu]-labeled estradiol displayed comparable tumor-targeting properties, suggesting little effect of the radionuclides on the tumor-targeting characteristics of the estradiol. There was no significant metabolism, with no evidence of degradation of the [68Ga]-estradiol observed. These findings validate the high stability obtained in plasma in vitro. The whole-body PET imaging studies displayed high uptake in the liver, intestines, and urinary bladder. Accumulation of radioactivity in the ER-positive MCF-7 tumor was somewhat low when compared to the background activity. The suitable pharmacokinetic properties and satisfactory tumor-targeting characteristics of the [68Ga/177Lu]-estradiol derivatives demonstrate their possible potential for effectively targeting estrogen receptor-positive breast carcinoma. The promising results from this research justify additional studies to assess the true efficacy of this novel radiolabeled estradiol derivative in breast cancer imaging and the potential monitoring of tumor response following therapy.
Conclusions
This study presents a novel method for preparing estradiol derivatives through solid-phase synthesis, along with the in vitro and in vivo assessment of new [68Ga/177Lu]-labeled estradiol derivatives in preclinical settings. The radiolabeled estradiol derivatives demonstrated significant metabolic stability. The binding affinity to the ER-positive breast cancer cell line was observed in the low nanomolar range, exhibiting both saturability and receptor specificity. Furthermore, the radiolabeled estradiol derivatives displayed rapid pharmacokinetics and effective tumor targeting in MCF-7 tumor xenograft models. These promising results may prove beneficial in the development of new and more potent estradiol-based radiopharmaceuticals aimed at efficiently targeting ER-positive tumors.
Materials and methods
General
All analytical grade chemicals and Fmoc-protected amino acids with appropriate side-chain protections were purchased from commercial sources and used without further purification. Estrone was purchased from Fisher Scientific, USA. DOTA-tris-(t-Bu ester) was bought from Macrocyclics, Dallas, TX. The structural identity of the newly synthesized estradiol was confirmed by mass spectrometry performed on Agilent 6125 single quadrupole liquid chromatography/mass spectrometry system (LC/MS) (Agilent Technologies, Santa Clara, CA, USA) using an eluent of 0.1% formic acid/29.95% H2O/ 69.95% CH3CN at a flow rate of 0.3 mL/min. Reversed-phase high-performance liquid chromatography (RP-HPLC) analyses were performed on a Shimadzu HPLC system (Shimadzu Corporation, Kyoto, Japan) fitted with a dual-wavelength UV-VIS detector (Shimadzu Corporation, Kyoto, Japan), a radioactivity detector (Bioscan, USA), and the Lauralite chromatogram analysis software (LabLogic Systems Ltd., Sheffield, UK). Radioactive samples were measured by using a γ-counter (Mucha, Raytest Isotopenmessgeräte GmbH, Straubenhardt, Germany).
The laboratory animals were received from the Jackson Laboratory, Bar Harbor, ME, USA. All the procedures and experiments involving laboratory animals were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) and Research Advisory Council (RAC; Project# 2250024) of the King Faisal Specialist Hospital and Research Centre (KFSH&RC). Animal housing, handling, and experimental procedures were conducted in the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) accredited laboratory animal facility at the KFSH&RC. Laboratory animal housing, care, and procedures were carried out according to the ARRIVE (Animal Research: Reporting of In Vivo Experiments) and IACUC guidelines.
Synthesis of acetic acid estradiol 3
Estrone 1 (3.3 mmol, 900 mg) was dissolved in DMF (N, N-dimethylformamide). To this, potassium carbonate (K2CO3, 550 mg, 1.2 equivalent) was added, and the mixture was stirred for 3 min at ambient temperature. Tert-butyl bromoacetate 2 (500 µL, 1.0 equivalent) was then added, followed by the addition of potassium iodide as a catalyst (KI, 65 mg, 0.1 equivalent). The reaction mixture was allowed to be stirred at ambient temperature for 24 h to ensure quantitative coupling. The solvents were removed on a rotary evaporator, the residue was diluted with ethyl acetate, and the organic layer was washed with 10% NaOH solution (2 × 20 mL) to remove unreacted estrone-OH. The organic layer was then separated and dried with anhydrous sodium sulfate for 30 min. The solvent was filtered off using a filter paper, and the filtrate was evaporated under reduced pressure to provide the alkylated product, which was used directly in the next step. The preparation of acetic acid estradiol is presented in Scheme S1.
Removal of the tert-butyl protecting group
The above-alkylated compound was dissolved in 2 mL of dichloromethane (DCM), and 2 mL of trifluoroacetic acid (TFA) was then added slowly. The resulting mixture was stirred at ambient temperature for 2 h. The solvents were removed on a rotary evaporator. To the residue, diethyl ether (10 mL) was added, and the product was precipitated as an off-white solid, which was collected by centrifugation. The purity of this acetic acid estradiol 3 product was checked by RP-HPLC analysis, and its structural identity by LC−MS analysis. Yield = 61%; LC-MS: m/z calcd for C20H24O4 328.4; found, 328.8 [M + H]+ [S3].
Synthesis of estradiol-lysine-DOTA
Solid-phase synthesis of the estradiol analog was started with Fmoc-Lys(ivDde)-wang resin using the peptide synthesis glass reaction vessel (Peptides International, Louisville, USA) (Scheme S2). For solid-phase synthesis, standard Fmoc (9-fluorenylmethyl-oxycarbonyl) chemistry was employed on a 0.2 mmol scale. The resin was soaked in DMF for 60 min and in DCM for 30 min. The solvent was drained, and the Fmoc protecting group of the lysine was removed by treating the resin with 20% (v/v) piperidine in DMF, followed by repeated washing with DMF and DCM (3 × 3 mL each). To attach estradiol to the wang resin, the acetic acid estradiol 3 was first preactivated at its α-carboxylic function using an activating reagent, HBTU (O-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) in the presence of a base DIEA (diisopropylethylamine) and then allowed to react for 60 min at ambient temperature with the resin-bound amino group of lysine to form an amide bond. The completion of the coupling was confirmed by the Kaiser ninhydrin test. At this point, the epsilon ivDde protecting group of the lysine was deprotected using 2% hydrazine monohydrate in DMF (3 × 3 mL, 7 min each), followed by standard washings with DMF and DCM (3 × 3 mL each) to facilitate the conjugation with DOTA bifunctional chelating agent.
The commercially available DOTA-tris-(t-Bu ester) (1,4,7,10-tetraazacyclododecane-1,4,7-tris-tert-butyl acetate-10-acetic acid) (2.5 equivalent) was first preactivated with HBTU (2.5 equivalent) in the presence of DIEA (5 equivalent) in DMF for 10 min before the conjugation with the resin. The coupling mixture was allowed to react for about 4 h at ambient temperature, and the completion of the reaction was confirmed by the negative Kaiser test. Finally, the DOTA-coupled estradiol derivative was cleaved from the resin along with other side-chain protecting groups by reacting the resin with a cleavage cocktail (~ 5 mL) of trifluoroacetic acid (TFA)/triisopropylsilane/water (95%: 2.5%: 2.5%, v/v) for 4 h at ambient temperature. The resin was removed by filtration, and TFA was evaporated by rotary evaporator. To the residue, cold diethyl ether was added to precipitate the product, which was isolated as a solid powder upon centrifugation thrice at 5000 RPM. The purity of the DOTA-estradiol product was checked by RP-HPLC, and its structural identity was confirmed by LC-MS analysis.
Yield = 40%; LC-MS: m/z calcd for C42H63N6O12 843.5; found, 844.5 [M + H]+ [S4].
Radiolabeling of DOTA-estradiol with 68Ga and 177Lu
Radiolabeling with 68Ga
The labeling procedure is basically as described previously26. Briefly, ~ 50 µg of DOTA-estradiol (1 mg/mL H2O/CH3CN (1:1 v/v) solution) was mixed with 200 µL of 2.5 M sodium acetate buffer. To this, 100 µL of EtOH was added. This was followed by the addition of [68Ga]GaCl3 in 0.05 M HCl (111–185 MBq; 3–5 mCi, 1 mL) (eluted from 68Ge–68Ga generator, ITG Germany), and adjusting the pH to ~ 4.5. The labeling mixture was then heated for 15 min at 90 °C to enhance labeling kinetics. The reaction mixture was cooled to room temperature and filtered through a 0.2‒µm pore syringe filter before HPLC analysis.
Radiolabeling with 177Lu
The labeling method is mostly as described previously26. Briefly, ~ 50 µg of peptide conjugate (1 mg/mL H2O/CH3CN (1:1 v/v) was mixed with ~ 300 µL of 0.5 M ammonium acetate buffer. To this, 40 µL gentisic acid (40 mg/mL aqueous solution), followed by ~ 100 µL of [177Lu]LuCl3 (74–148 MBq; 2–4 mCi), dissolved in 0.04 M HCl, was added. The reaction mixture with a final pH of 4.5 was heated at 90 °C for 30-min to accelerate labeling kinetics. After cooling, the preparation was filtered through a 0.2‒µm pore syringe filter before HPLC analysis.
A non-radioactive gallium complex, natGa-DOTA-estradiol, was prepared by reacting DOTA-estradiol (3 mg, 0.003 mmol) with Ga(III)Cl3 (1.0 g, 2 equivalent) dissolved in 400 µL of 0.05 M HCl. To this, 400 µL of 1.0 M sodium acetate buffer (pH 5.0) was added. The reaction mixture was heated for 60 min at 90 °C and analyzed by HPLC.
HPLC analysis
Reversed-phase HPLC analyses were carried out following our previously reported method26,27. In short, the HPLC analysis and purification were performed on a Shimadzu HPLC system (Shimadzu, Japan) using a Phenomenex Luna C18 reversed-phase column (5 μm, 150 × 4.6 mm). For all HPLC experiments, a gradient system of 0.1% (v/v) TFA in water (solvent A) and 0.1% (v/v) TFA in CH3CN (solvent B) at a flow rate of 1 mL/min was used. The HPLC gradient elution was started with a solvent composition of 95% A and 5% B from 0 to 2 min, followed by a linear gradient of 95% A and 5% B to 5% A and 95% B over 25 min. The gradient remained at this position for 3 min before switching back to the initial settings of 95% A and 5% B for another 5 min. The major peak of the radiolabeled estradiol was collected, and the organic solvent was then slowly evaporated under a stream of nitrogen gas. Radiochemical purity was estimated by evaluating the radioactivity peak eluted and calculating the area under the peak (ROI). The HPLC-purified estradiol product was reconstituted in sterile saline and used for in vitro and in vivo assays.
Octanol/water partition coefficient
[68Ga/177Lu]-DOTA estradiol (25 µL, 5 µCi) was added into a glass tube containing 1 mL each of n-octanol and double-distilled water (pH = 7.4). The mixture was vortexed for 2 min and then centrifuged (5000 RPM, 5 min) to ensure the complete separation of layers. Three 100 µL aliquots from both the octanol and water layers were aspirated, and radioactivity was determined using a γ-counter. The octanol-water partition coefficient (Log Po/w) was calculated as Log(cpm in octanol/cpm in water). The experiment was performed in triplicate.
Stability in human plasma
Human plasma was obtained from the institutional blood bank from healthy volunteer blood donors (with consent). The metabolic stability of the [68Ga/177Lu]-DOTA estradiol derivatives was determined following a method essentially as described previously26,27. In summary, the [68Ga/177Lu]-DOTA estradiol (50 µL, 100 µCi) was mixed with plasma (400 µL) and incubated in duplicate at 37 °C for up to 4 h. After incubation, the plasma proteins were precipitated using a mixture of CH3CN/EtOH (1:1 v/v, 400 µL). The supernatant layer was collected by centrifugation (5000 RPM, 5 min), filtered through a 0.2-µm Millex LG filter, and analyzed by radio-HPLC to assess the radioestradiol stability.
Cell line preparation
The cells were prepared essentially as previously described27. In brief, the ER-positive MCF-7 breast cancer cell line (American Type Culture Collection, Rockville, USA) was grown as monolayers at 37 °C in a humidified atmosphere containing RPMI-1640 culture media with 10% fetal bovine serum (FBS) in the tissue culture flasks. Twenty-four hours before conducting the tumor implantation, the media was replaced with RPMI-1640/10% FBS. The cells were grown to 80−90% confluency and harvested by trypsinization. After centrifugation, approximately 50 million cells were suspended in 2 mL PBS. For inoculation per nude mouse, approximately 5 million MCF-7 cells in 200 µL sterile PBS were implanted subcutaneously into each nude mouse. Once tumor growth became sufficient (~ 1 cm), tumor uptake and organ biodistribution were carried out as described below.
In vitro cell binding
The binding of the [68Ga/177Lu]-DOTA estradiol to ER-positive MCF-7 human breast cancer cell line (obtained from the ATCC, Rockville, MD, USA) was carried out essentially as described previously26. Briefly, six different concentrations of the DOTA-estradiol, ranging from 1.0 to 200 nM (prepared from the serial dilutions of HPLC-purified compound) mixed with a fixed amount of 200,000 cells (in 200 µL phosphate-buffered saline, PBS) at room temperature (21–23 °C), with slow shaking in duplicate for 60 min. The initial concentration of DOTA-estradiol was determined following a known HPLC technique with simultaneous detection by UV absorbance27. Incubation was terminated by dilution with cold PBS (200 µL), followed by centrifugation. The supernatant was collected, and cell pellets were rapidly washed with cold PBS to remove any unbound radioestradiol. Radioactivity in the cell pellet (total bound) and the supernatant (unbound) was measured using a γ-counter. Nonspecific binding was determined in the presence of approximately 200-fold molar excess of unlabeled DOTA-estradiol. Specific binding is calculated by subtracting the non-specifically bound radioactivity from that of the total binding. The dissociation constant (Kd) is calculated using a plot of cell-bound activity versus the concentrations of the radioligand using the GraphPad Prism software version 6 (GraphPad Software Inc., San Diego, CA, USA). The experiment was performed in triplicate.
Internalization
Internalization experiments were performed as described previously26. In brief, approximately 200,000 MCF-7 breast cancer cells were incubated with [177Lu]-DOTA estradiol for 60 min at 37 °C. After incubation, cells were separated by centrifugation and washed twice with 1 mL PBS. Subsequently, cells were washed twice with 1 mL of acidic buffer (0.02 M sodium acetate in saline, pH 5.0)28 for 10 min at room temperature to remove the surface-bound radioactivity. The cells were washed again twice with 1 mL of ice-cold PBS and lysed with 0.5 mL of 1 M NaOH. The amount of cell surface-bound (acid-wash) and intracellular radioactivity (acid-resistant) was determined using a γ-counter.
Dose preparation
For in vivo biodistribution studies, doses were prepared by dissolving the HPLC-purified [68Ga/177Lu]-DOTA estradiol in sterile saline to a concentration of ~ 100 µCi/mL. For PET animal imaging studies, doses were prepared by dissolving the [68Ga]-DOTA estradiol in saline to ~ 1.5 mCi/mL. For the receptor-blocking experiment, DOTA-estradiol was dissolved in a dose solution of ~ 2 mg/mL. The resulting dose solution was filtered with a 0.2-µm Millex LG filter before being injected into animals. Each animal was injected with ~ 100 µL of the dose solution.
In vivo tumor uptake study
Approval from the Institutional Animal Care and Use Committee was obtained for the animal protocol used. In addition, strict international regulations govern the safe and proper use of laboratory animals employed during animal experiments. The animal study is reported per ARRIVE guidelines. Mice used for this research were housed under controlled conditions of 12 h of light/dark cycles, temperature (~ 20–23 °C), humidity (40–60%), and free access to regular commercial mouse feed and water. In vivo tumor uptake was conducted following the method reported previously26,27. In short, tumor uptake and tissue biodistribution studies were carried out in female nude mice (n = 5 per time point, body mass 20–25 g) at 45 min and 2.5 h for [68Ga]-DOTA-estradiol and 1 h and 4 h for [177Lu]-DOTA-estradiol. Each animal was administered with the HPLC-purified [68Ga]- or [177Lu]-labeled DOTA-estradiol, ∼0.001 nmol, 100 µL, 20–25 µCi, via lateral tail vein injection. At the specified time points, the ER-positive xenografted nude mice were sacrificed by cervical dislocation. A fraction of the blood (∼100 µL) was collected from cardiac puncture. Urine was also collected and measured with the bladder contents. The tumor was dissected, and major organs, such as the lungs, pancreas, stomach, liver, heart, kidneys, and intestines, were isolated, weighed, and measured for radioactivity. The percent of the injected dose or injected activity per gram (% ID/g or % IA/g) in the tumors and body organs was determined by using a custom-designed Quattro Pro X9 spreadsheet (Corel Corporation, USA). The uptake values of the radioestradiol are expressed as the % IA/g of tissue/organ. The radioactivity in the urine with bladder contents is shown as the % IA/organ. To calculate the injected activity, a standard (20-fold dilution of injected activity) was prepared and counted together with animal tissue samples.
For the ER blocking study, an additional group of mice was injected with the [68Ga]-DOTA-estradiol premixed with unlabeled DOTA-estradiol (~ 0.23 nmol, 50 µL, 100 µCi). In vivo biodistribution and blocking studies were performed at 2.5 h p.i. as stated before.
Metabolism studies
To determine the radio-metabolites of the [68Ga/177Lu]-DOTA-estradiol, an aliquot of the urine sample was collected by manual void at the time of sacrifice and mixed with an equal volume of CH3CN. The mixture was centrifuged at 5000 RPM for 5 min. The resulting supernatant was collected and passed through a 0.20 μm Millex-LG filter unit to remove any precipitate and analyzed by radio-HPLC.
Small animal PET imaging
Micro PET imaging was conducted as described previously with some modifications29. To find out the tumor-targeting potential of the [68Ga]-DOTA-estradiol (∼0.001 nmol, 100 µL, ∼150 µCi) was administered through the tail vein injection into a nude mouse carrying MCF-7 tumor xenografts. After 45 min p.i., the mouse was sedated under anesthesia and placed in the Nano-PET/CT scanner (Mediso, Hungary), with continuous 2% isoflurane in oxygen flow and whole-body imaged for 15 min in the prone position. Images were then reconstructed by three-dimensional ordered subsets, and the acquired data were analyzed by InterView FUSION software (Mediso, Hungary). Following imaging, animals were dissected, and quantitative biodistribution was performed to validate the findings of the PET imaging.
Statistical analysis
Experimental data are represented as mean ± S.D., where appropriate. For data evaluation, mean values were compared using the Student’s t-test (GraphPad Software, San Diego, CA), and a probability value (P) ˂0.05 was considered statistically significant.
Data availability
Data is available from the corresponding author upon request.
References
Lau, J. et al. Insight into the development of PET radiopharmaceuticals for oncology. Cancers 12, 1312 (2020).
Fani, M., Maecke, H. M. & Okarvi, S. M. Radiolabeled peptides: valuable tools for the detection and treatment of cancer. Theranostics 2, 481–501 (2012).
Gradishar, W. J. et al. NCCN Clinical practice guidelines in oncology. Breast cancer, version 5 2020.. https://www.nccn.org/professionals/physician_gls/default.aspx. (2020).
Allison, K. H. et al. Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP guideline update. J. Clin. Oncol. 38, 1346–1366 (2020).
Blamey, R. W. et al. ONCOPOOL – A European database for 16,944 cases of breast cancer. Eur. J. Cancer. 46, 56–71 (2010).
Cardoso, F. et al. Characterization of male breast cancer: results of the EORTC 10085/TBCRC/BIG/NABCG. International male breast cancer program. Ann. Oncol. 29, 405–417 (2018).
Thompson, E. B. et al. Steroid Receptors and the Management of Cancer. Vol. 1, 1–248 (CRC Press, 1979).
Allegra, J. C. Distribution, frequency, and quantitative analysis of estrogen, progesterone, androgen, and glucocorticoid receptors in human breast cancer. Cancer Res. 29, 1447–1454 (1979).
Kiesewetter, D. O. et al. Preparation of four Fluorine-18 labeled estrogens and their selective uptakes in target tissues of immature rats. J. Nucl. Med. 25, 1212–1221 (1984).
Nayak, T. K. et al. Preclinical development of a neutral, estrogen receptor–targeted, tridentate 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative for imaging of breast and endometrial cancers. J. Nucl. Med. 49, 978–986 (2008).
Bafaloukos, D. et al. Neo-adjuvant therapy in breast cancer. Ann. Oncol. 16, ii174–ii181 (2005).
Veronesi, U. et al. Breast cancer. Lancet 365, 1727–1741 (2005).
van Kruchten, M. et al. PET imaging of oestrogen receptors in patients with breast cancer. Lancet Oncol. 14, e465–e475 (2013).
Friedel, A., Prante, O. & Maschauer, S. Radiosynthesis and preclinical evaluation of 18F-Labeled estradiol derivatives with different lipophilicity for PET imaging of breast cancer. Cancers 16, 2639 (2024).
Xu, D. et al. 18F-labeled estradiol derivative for targeting estrogen receptor-expressing breast cancer. Nucl. Med. Biol. 59, 48–55 (2018).
Chauhan, K. et al. Bivalent approach for homodimeric estradiol based ligand: synthesis and evaluation for targeted theranosis of ER(+) breast carcinomas. Bioconjug. Chem. 27, 961–972 (2016).
Xia, X. et al. 99mTc-labeled estradiol as an estrogen receptor probe: Preparation and preclinical evaluation. Nucl. Med. Biol. 43, 89–96 (2016).
Xu, D. et al. Radioiodinated estradiol dimer for estrogen receptor targeted breast cancer imaging. Chem. Biol. Drug Des. 96, 1332–1340 (2020).
Katzenellenbogen, J. A. The quest for improving the management of breast cancer by functional imaging: the discovery and development of 16α-[18F]fluoroestradiol (FES), a PET Radiotracer for the estrogen receptor, a historical review. Nucl. Med. Biol. 92, 24–37 (2021).
Katzenellenbogen, J. A., Welch, M. J. & Dehdashti, F. The development ofestrogen and progestin radiopharmaceuticals for imaging breast cancer. Anticancer Res. 17, 1573–1576 (1997).
Fluoroestradiol, F. 18 – a molecular marker becomes commercially available for PET imaging in metastatic breast cancer. Mol. Imaging Siemens. https://www.siemens-healthineers.com.
CERIANNA™ (fluoroestradiol F-18) Injection, for intravenous use Initial U.S. Approval: 2020. Reference ID: 4610145-Accessdata.fda.gov.
Skaddan, M. B., Wust, F. R. & Katzenellenbogen, J. A. Synthesis and binding affinities of novel Re-containing 7α-substituted estradiol complexes: models for breast cancer imaging agents. J. Org. Chem. 64, 8108–8121 (1999).
Szymiczek, A., Lone, A. & Akbari, M. R. Molecular intrinsic versus clinical subtyping in breast cancer: A comprehensive review. Clin. Genet. 99, 613–637 (2021).
Yao, J. et al. Optimization of small molecule degraders and antagonists for targeting estrogen receptor based on breast cancer: current status and future. Front. Pharmacol. 14, 1225951 (2023).
Okarvi, S. M. Synthesis: Radiolabeling and in vitro and in vivo characterization of a technetium-99m-labeled alpha-M2 peptide as a tumor imaging agent. J. Pept. Res. 63, 460–468 (2004).
Okarvi, S. M. & Al-Jammaz, I. A. Convenient and efficient total solid-phase synthesis of DOTA-functionalized tumor-targeting peptides for PET imaging of cancer. EJNMMI Res. 9, 88 (2019).
Breeman, W. A. et al. Preclinical comparison of 111In-labeled DTPA- or DOTA-bombesin analogs for receptor-targeted scintigraphy and radionuclide therapy. J. Nucl. Med. 43, 1650–1656 (2002).
AlHokbany, N. et al. Development of new copper-64 labeled rhodamine: a potential PET myocardial perfusion imaging agent. EJNMMI Radiopharm Chem. 23, 19 (2022).
Acknowledgements
The authors would like to thank Dr. Monther Alalwan for providing the cells; Celestina Maccuto for animal experiments, and Dr. Falah Mohanna for assistance in the induction of cancer cells in mice. Yousef Maliki for performing the imaging study. The support of the Research and Innovation Administration is greatly acknowledged.
Funding
The study was carried out within the scientific scope of the organizations and supported by departmental funding.
Author information
Authors and Affiliations
Contributions
S.M.O. planned and designed the experiments and interpreted the results. S.M.O. performed the experiments and wrote the paper. All helping technicians reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Okarvi, S.M. A novel solid phase synthesis of an estradiol derivative and preclinical evaluation for targeting estrogen-receptor positive breast cancer. Sci Rep 15, 30143 (2025). https://doi.org/10.1038/s41598-025-15367-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15367-0
