
Abstract
In the present work, new quinolone-2-thio-acetamide-propane hydrazide-benzimidazole derivatives 12a-o were assigned as potent anti-diabetic agents that targeting α-glucosidase and α-amylase as two important targets in treatment of type 2 diabetes. General scaffold of these compounds was designed based on the reported potent α-glucosidase and α-amylase inhibitors and derivation was performed in acetamide moiety. In vitro evaluation of the new compounds 12a-o demonstrated that most of the synthesized compounds were more potent than standard inhibitor acarbose against α-glucosidase while all these new compounds were more potent than acaerbose against α-amylase. The most potent compound against both studied enzymes was compound 12n that was a 4-fluorophenylacetamide derivative. This compound was 5 and 23.8 folds more potent than acarbose against α-glucosidase and α-amylase, respectively, and with excellent binding energies in comparison to acarbose attached to active sites of these enzymes. Molecular dynamics and pharmacokinetic studies of compound 12n was also performed.
Similar content being viewed by others
Introduction
Diabetes is a chronic metabolic disease that caused by insulin deficiency (either in insulin production or action, or both) and characterized by hyperglycemia1. Hyperglycemia can lead to serious long-term complications, including cardiovascular disease, nerve damage, and failure of the kidneys, nerves, and eyes2. It is estimated that by 2045, more than 700 million people will have diabetes. Diabetes can be classified into two main categories: type 1 and type 2. Type 2 diabetes accounts for about 90% of all diabetes cases and is caused by insulin deficiency3. Although no definitive treatment has been proposed to reduce the metabolic disorders associated with it, one of the most effective methods to manage type 2 diabetes is controlling blood sugar levels after meals. This can be achieved by inhibiting the carbohydrate-hydrolyzing enzymes α-amylase and α-glucosidase4,5. α-Amylase (EC 3.2.1.1) is one of the most well-known amylolytic enzymes that found in many tissues, prominently in pancreatic juice and saliva6. This enzyme known as a 4-α-D-glucan gluconohydrolase and is responsible for hydrolyzing the α-bonds of large polysaccharides like starch and glycogen into shorter chains such as dextrin and maltose7. α-Glucosidase known systematically as α-D-glucoside glucohydrolase and is located in the brush border of enterocytes in the small intestine8. This enzyme hydrolyzes α-1→4 glucosidic linkages of disaccharides and oligosaccharides to release monosaccharides (α-D-glucose anhydrofructose)9. For diabetic patients, α-glucosidase inhibitors like acarbose, voglibose, and miglitol are considered as safe drugs, however, some studies have shown that these drugs can be associated with gastrointestinal complications, including diarrhea, bloating, and abdominal cramping10. The potential side effects of α-glucosidase inhibitors have prompted medicinal chemists to design new efficient, safe, and more potent α-glucosidase inhibitors with fewer side effects.
Over the past decade, computational (in silico) methodologies, particularly molecular docking, virtual screening, and pharmacophore modeling, have become fundamental tools in rational drug design and modern drug discovery. These approaches enable rapid, cost-effective prediction of biological activity, target binding affinities, and structure-activity relationships, with particular utility in studying natural products and semi-synthetic compounds11,12,13,14,15. Recent advances demonstrate how integrating these in silico techniques with experimental validation (in vitro/in vivo) significantly enhances the discovery and optimization of novel therapeutics16,17,18. This strategy has proven highly effective in the development of heterocyclic scaffolds and allows for the rational design of compounds with enhanced biological activity.
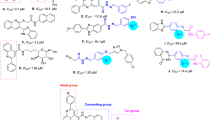
Quinoline is a bicyclic N-heterocycle that exhibits various biological effects such as antifungal, antimalarial, antibacterial, anthelmintic, anticancer, anticonvulsant, anti-inflammatory, and analgesic properties19. Furthermore, very synthetic α-glucosidase inhibitors derived from quinoline have been reported20,21,22. For example, compounds A-C are derivatives of quinolone that demonstrated high inhibitory activity against α-glucosidase23,24,25. As can be seen in Fig. 1, in the design of compounds A-C, quinoline was combined with 2-thio-acetamide derivatives. On the hand, benzimidazole is an interest pharmacophore in the design of α-glucosidase and α-amylase inhibitors26,27,28. As can be seen, this pharmacophore was also seen in α-glucosidase inhibitors C (Fig. 1)25. Recently, our research group reported benzimidazole-propane hydrazide derivatives D with dual inhibitory activities against α-glucosidase and α-amylase29. It is worth mentioning that the hydrazide group is also widely used in the design of α-glucosidase and α-amylase inhibitors30.

Reported α-glucosidase (α-amylase) inhibitors containing quinoline and/or benzimidazole pharmacophores.
Therefore, based on the depicted design strategy in Fig. 2, we designed quinolone-2-thio-acetamide-propane hydrazide-benzimidazole scaffold and fifteen derivatives 12a-o synthesized of it. In vitro and in silico evaluations of these derivatives were performed against α-glucosidase and α-amylase.

Rational design for the quinolone-2-thio-acetamide-propane hydrazide-benzimidazole derivatives 12a–o as new anti-diabetic agents.
Results and discussion
Chemistry
New quinolone-2-thio-acetamide-propane hydrazide-benzimidazole derivatives 12a-n were synthesized according to Scheme 1. This scheme showed that for the synthesis of title compounds, two starting materials 3-(1 H-benzo[d]imidazol-2-yl)propanehydrazide (5) and 2-((3-formylquinolin-2-yl)thio)acetamide derivatives 11a-n were needed. Compound 5 was synthesized in three steps: 1) in the first step, succinic anhydride (1) reacted with o-phenylenediamine (2) in xylene to give 3-(1 H-benzo[d]imidazol-2-yl)propanoic acid (3), 2) in the second step, compound 3 in the presence of sulfuric acid at reflex condition converted to methyl 3-(1 H-benzo[d]imidazol-2-yl)propanoate (4), and 3) in the third step, compound 5 was obtained of reaction between compound 4 and hydrazine. On the other hand, compounds 11a-n were also synthesized in three steps: (1) N, N-dimethylformamide (6) reacted with 2-chloro-N-phenylacetamide (7) in the presence of phosphoryl chloride (POCl3) to give 2-chloroquinoline-3-carbaldehyde (8), (2) the latter compound in the presence of sodium sulfide (Na2S) converted to 2-mercaptoquinoline-3-carbaldehyde (9), and (3) compound 9 reacted with acetamide derivatives 10a-n to give 2-((3-formylquinolin-2-yl)thio)acetamide derivatives 11a-n. In the final step, compound 5 reacted with derivatives 11a-n in the presence of PTSA to give target compounds 12a-n.

Synthetic procedure for preparation of new compounds 12a-n.
In vitro α-glucosidase and α-amylase Inhibition assays of the new derivatives 12a–o
All the synthesized compounds 12a-o were evaluated against α-glucosidase and α-amylase as two important targets for treatment of diabetes and the obtained results were listed in Table 1.
α-Glucosidase inhibition assay demonstrated that all the synthesized derivatives 12a-o were active against this enzyme and more than half of them (twelve derivatives) inhibited the target enzyme better than the standard drug acarbose. The range of IC50 values of compounds 12a-o was 10.36 to 70.11 nM and IC50 value of acarbose was 52.26 nM. Therefore, the most potent compound against α-glucosidase (compound 12n) was 5 fold more potent that standard inhibitor.
In term of anti-amylase activity, all the synthesized compounds 12a-o with IC50 values 1.71–22.21 nM were more potent than standard inhibitor acarbose with IC50 value 40.74 nM. Among the new compounds 12a-o, compound 12n with inhibitory activity 23.8 fold more than acarbose, was the most potent entry.
As can be seen in Scheme 1, most synthesized compounds were derivatives of aniline. Used substituents in the aniline derivatives 12a-n were showed in Table 1 and structure-activity relationships (SARs) of these compounds in performed assays were schematically sowed in Scheme 2. It should be noted that, we also synthesized the benzylamine derivative 12o. This compound did not show significant activities against α-glucosidase but were good inhibitors against α-amylase.
Survey on SARs of N-phenylacetamide derivatives 12a-n against α-glucosidase demonstrated that the most potent compounds were 4-fluoro derivative 12n, 2,4-dichloro derivative 12j, and 2-fluoro derivative 12 m, respectively (Table 1 and Scheme 2). The fourth potent compound against α-glucosidase was 3-chloro derivative 12 h. Movement of chloro substituent of 3-position to 2 or 4-position, in the cases of compounds 12 g and 12i, respectively, created a moderate decrease in inhibition effect.
The order of anti-α-glucosidase activity in derivatives with a substituent on 4-position of phenyl ring of N-phenylacetamide moiety was F > Nitro > Cl > OMe > Me and this order demonstrated that electron withdrawing groups have better activities of electron donating groups. Moreover, as can be seen in Scheme 2, a general review on SAR shows that electron withdrawing substitutions have better effects on anti-α-glucosidase activity in comparison to electron donating groups. In this regards, all the N-phenylacetamide derivatives with inhibitory activity less than acarbose, were derivatives of methyl (2,3-dimethyl derivative 12e, 3-methyl derivative 12b, and 4-methyl derivative 12c, respectively). Moreover, un-substituted compound 12a and 2,4-dimethyl derivative 12f were negligibly more potent than acarbose.
SARs of compounds 12a-n in anti-α-amylase assay showed that, like anti-α-glucosidase assay, the most potent compounds were compounds 12n, 12j, and 12 m, respectively, and order of activity in 4-substituented derivatives was F > Nitro > Cl > OMe > Me. The forth potent compound against α-amylase was 4-nitro derivative 12k. Adding a methyl substituent on 2-position of compound 12k, as in the case of compound 12 L, decreased anti-α-amylase activity to two folds. As can be seen Table 1, chlorine derivatives showed good effects against α-amylase with fallowing order: 2,4-dichloro (the second potent compound 12j) > 3-Cl (the fifth potent compound 12 h) > 2-Cl (the sixth potent compound 12 g) > 4-Cl (the seventh potent compound 12i). Un-substituted compound 12a was also a good α-amylase inhibitor and introduction of electron donating substituents 2,4-dimethyl, 3-methyl, 4-methoxy, 2,3-dimethyl, and 4-methyl on did not improve anti-α-amylase activity in comparison to un-substituted derivative 12a.

Diagram of SARs of aniline derivatives 12a-n in the anti-α-glucosidase and anti-α-amylase assays.
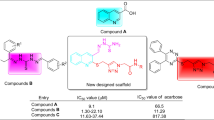
Comparison of anti-α-glucosidase and anti-α-amylase activities of new compounds 12a-n with used template compounds A and B were shown in Scheme 315, 16. As can be seen in Scheme 3, our new compounds 12a-o were significantly more potent than template compounds B in term of inhibitory activity against both digestive enzymes α-glucosidase and α-amylase. In contrast, our new compounds were less active than template compounds A against α-glucosidase.

Comparison of new compounds 12a-o with used template compounds A and B.
Docking study
We conducted molecular docking studies to identify interaction modes of compound 12n and α-glucosidase and α-amylase because this compound was the most potent compound against both these enzymes.
The 3D and 2D interaction modes of compound 12n within the α-glucosidase’s active site was showed in Fig. 3. According this figure, compound 12n formed two hydrogen bonds with the active site residues Asp616 and Ala284. This compound also established a halogen interaction via fluorine atom with Asp404, two electrostatic interactions with Asp282 and Asp616, and two π-Sulfur interactions with Met519 and Trp481. Additionally, compound 12n formed several hydrophobic interactions with residues Trp481, Trp376, Leu678, and Ala555.

3D (a) and 2D (b) interaction modes of compound 12n within the active site pocket of α-glucosidase (https://discover.3ds.com/).
Docking study on compound 12n in the active site of α-amylase demonstrated that this compound formed two hydrogen bonds with Arg195 and Asp300, two halogen interactions through the fluorine atom with residues Asp197 and His299, and four π-π interactions with residues His201 (two interaction), Try62, and Trp59. This compound also formed hydrophobic interactions with residues Ile235, Lys200, Leu165, and Leu162 (Fig. 4).

3D (a) and 2D (b) interaction modes of compound 12n within the active site of α-amylase (pdb code: 4w93) (https://discover.3ds.com/).
The binding energy values of compound 12n in the active sites of α-glucosidase and α-amylase were − 8.13 and − 8.80 kcal/mol, respectively, while binding energy values of acarbose in the α-glucosidase’s active site was − 7.34 and α-amylase in the α-amylase’s active site was − 7.17 kcal/mol. The latter in silico findings are in agreement with in vitro data.
Molecular dynamics (MD) studies
The biological results showed that compound 12n has the greatest potential to inhibit α-glucosidase and α-amylase. MD studies were performed on the α-glucosidase-compound 12n and α-amylase-compound 12n complexes in term of stability, flexibility, and intermolecular interactions during 100 ns. To assess the stability of the latter complexes, the root mean square deviation (RMSD) and radius of gyration (Rg) for all structures in the pathway were calculated. Additionally, the root mean square deviation (RMSF) of the backbone atoms in α-glucosidase and or α-amylase and ligand atoms were also calculated to assess the residual flexibility and the flexibility of the ligand atoms. The graphs of these calculations were generated during the simulation time (Figs. 5 and 6). Figures 5a and 6a show the RMSD calculation results for α-glucosidase and α-amylase, respectively. As indicated in Fig. 5a, the RMSD of α-glucosidase remained consistently low, below 0.25 nm throughout the entire simulation period, demonstrating the stability of the protein-ligand structure during the 100 ns simulation. The average RMSD values of α-glucosidase–compound 12n complex and the α-glucosidase–acarbose complex were 0.18 nm and 0.20 nm, respectively. The RMSDs of the two systems (Fig. 5a) remained stable in the last 36 ns for the α-glucosidase-compound 12n complex and α-glucosidase-acarbose complex, stabilizing around an average value of 0.19 nm and 0.21 nm, respectively. Figure 6a shows the RMSD calculation results for α-amylase. The RMSD of α-amylase was shown to be lower than 0.2 nm, reflecting the stability of the protein-ligands interactions. The average RMSD values of α-amylase– compound 12n complex and the α-amylase–acarbose complex were 0.14 nm, and in the last 17 ns for the α-amylase-compound 12n complex and in the last 33 ns for α-amylase-acarbose complex, stabilizing around an average value of 0.15 nm and 14 nm, respectively (Fig. 6a). The RMSF values for the atoms of acarbose and compound 12n were also calculated. The oscillation of ligand atoms is shown in Figs. 5b and 6b. All atoms of compounds 12n and acarbose exhibited RMSF values of less than 3 Å. The loops provide several effective hydrophobic interactions with binding site residues, confining atomic fluctuations. The Rgs of α-glucosidase and α-amylase were calculated to evaluate the protein’s compactness and stability during the simulation time (Figs. 5c and 6c, respectively). Figure 5c shows that the Rg values of α-glucosidase fluctuated between 2.82 and 2.88 nm for both complexes during the simulation, indicating minor changes in protein compaction and stability of the α-glucosidase structure during the simulation period. The average Rg for both α-glucosidase complexes with acarbose or compound 12n was determined to be 2.85 nm. On the other hand, Fig. 6b shows that the value of Rg for α-amylase was between 2.30 and 2.36 nm for both complexes during the simulation and the average Rg for both α-amylase complexes with acarbose or compound 12n was determined to be 2.32 nm. The number of hydrogen bonds between the ligands and enzymes (α-glucosidase and α-amylase) is shown in Figs. 5d and 6d. During the simulation, the number of hydrogen bonds between the ligands and enzymes (α-glucosidase and α-amylase) fluctuated. For both of the enzymes (α-glucosidase and α-amylase)–acarbose complex, the number of hydrogen bonds mainly varied between 2 and 8, indicating a strong complex. According to docking studies, compound 12n forms two hydrogen bonds with amino acids in the binding site of enzymes (α-glucosidase and α-amylase) (Figs. 3 and 4) while MD simulations showed that the number of hydrogen bonds in the enzymes- compound 12n complex varies between 2 and 4.

(a) Superimposed RMSD, (b) Superimposed RMSF, (c) time dependence of Rg graph, and (d) the number of hydrogen bonds of compound 12n (mustard) and acarbose (navy) in complex with α-glucosidase (http://www.gromacs.org).

(a) Superimposed RMSD, (b) Superimposed RMSF, (c) time dependence of Rg graph, and (d) the number of hydrogen bonds of compound 12n (purple) and standard inhibitor acarbose (orange) in complex with α-amylase (http://www.gromacs.org).
In Silico druglikeness evaluation and prediction of ADMET profile
Druglikeness evaluation and ADMET prediction of acarbose and compound 12n were studied by PreADMET as a credible online server. The obtained data were listed in Table 2. A general rule for evaluation of druglikeness is Lipinski ‘Rule of five’ and the obtained results of the later software demonstrated that compound 12n followed of this rule while acarbose violated. Prediction of pharmacokinetic properties of compound 12n demonstrated that permeability of this compound to Caco2 cell, like acarbose, is poor while compound 12n, unlike acarbose, had high human intestinal absorption (HIA). Permeability of compound 12n and acarbose to blood brain barrier (BBB) and skin is in the acceptable range. In term of mutagenicity, both studied compounds are mutagen. In term of carcinogenicity, compound 12n has not carcinogenic effect on mouse and has this effect on rat while acarbose has carcinogenic effect on mouse and has not carcinogenic effect on rat. In term of cardiotoxicity (hERG inhibition), acarbose is ambiguous while compound 12n has high risk.
Bioavailability of compound 12n was also evaluated by two other online servers SwissADME and ADMETlab 2.0 (Fig. 7). Left radars in Fig. 7 were obtained by SwissADME server. In these radars presence of a compound in the pink region, as an optimal property range, is favorable. Therefore, this radar in the case of compound 12n and acarbose, showed that our new compound has better bioavailability in compression to acarbose. Right radars in Fig. 7 were depicted by ADMETlab 2.0 server. Bioavailability in these radars was determined by the presence of the compound in the colored area. Therefore, as can be seen Fig. 7, compound 12n has better bioavailability in compression to acarbose.

Bioavailability radars for compound 12n and acarbose (left radars were prepared by SwissADME (https://www.swissadme.ch/) server and right radars were prepared by ADMETlab 2.0 server (https://admetmesh.scbdd.com/).
Conclusion
In this study, a new series of quinolone-2-thio-acetamide-propane hydrazide-benzimidazole derivatives 12a-o was designed based on molecular hybridization, considering the structures of strong α-glucosidase and α-amylase inhibitors. After synthesizing these compounds, their effects against the latter enzymes were evaluated under laboratory conditions and the obtained results were compared to acarbose as a standard inhibitor. These results showed that most of our new compounds were more potent than acarbose against α-glucosidase while all the new compounds 12a-o were more potent than acarbose. Among the synthesized compounds, compound 12n was the most potent compound against both target enzymes. In silico docking and dynamics studies demonstrated that compound 12n as well interacted with the active sites of α-glucosidase and α-amylase and formed stable complexes with active sites of these enzymes. Compound 12n also had good pharmacokinetic properties based on in silico studies.
Experimental
Synthesis of 3-(1 H-benzo[d]imidazol-2-yl)propanehydrazide (5)
A mixture of succinic anhydride (1, 20 mmol) and o-phenylenediamine (2, 20 mmol) in xylene (100 mL) was stirred at reflux condition for 24 h. After that, xylene was evaporated under reduced pressure, the obtained residue was dissolved in ethyl acetate (100 ml), and the ethyl acetate phase was washed with water (4 × 100 ml) and brine (100 ml). Then, the organic phase was separated and dried over Na2SO4 and the ethyl acetate was evaporated under reduced pressure to give pure 3-(1 H-benzo[d]imidazol-2-yl)propanoic acid (3). To compound 3 (20 mmol), H2SO4 (10 mol%), and methanol (50 mL) were added and obtained mixture was heated at reflux condition for 16 h. Then, cold water was added to the latter mixture and obtained precipitate was dried at 60 °C to obtain 3-(1 H-benzo[d]imidazol-2-yl)propanoate (4). A mixture of compound 4 (20 mmol) and hydrazine (40 mmol) in ethanol (50 mL) was heated at reflux condition for 8 h. After that, water was added to the reaction mixture and observed participate was filtered and dried at 60 °C to give pure 3-(1 H-benzo[d]imidazol-2-yl)propanehydrazide (5).
General procedure for the synthesis of 2-((3-formylquinolin-2-yl)thio)acetamide derivatives 11a-o
N, N-dimethylformamide (DMF, 6) as both solvent and reagent reacted with 2-chloro-N-phenylacetamide (7, 20 mmol) and POCl3 (20 mmol) at 85 °C for 12 h. Then, cold water was added to it and obtained participate was filtered and dried at 60 °C to give pure 2-chloroquinoline-3-carbaldehyde (8). In the next step, compound 8 (20 mmol) and Na2S (20 mmol) in DMF (40 mL) were stirred at room temperature (RT) for 2 h. After this period, water was added to the reaction mixture and 2-mercaptoquinoline-3-carbaldehyde (9) was separated by filtration. Compound 9 (1 mmol), acetamide derivatives 10a-o (1 mmol), and K2CO3 (1.5 mmol) in DMF (10 mL) were stirred at room temperature for 5 h and after this time, by adding water, precipitates 11a-o were obtained.
General procedure for the synthesis of quinolone-2-thio-acetamide-propane hydrazide-benzimidazole derivatives 12a-o
n the final step, a mixture of compound 5 (1 mmol), 2-((3-formylquinolin-2-yl)thio)acetamide derivatives 11a-o, and PTSA (1 mmol) in ethanol (10 mL) was stirred at 80 °C for 24 h. After that, cold water was added to this reaction mixture and precipitates of target compounds 12a-o were formed that after filtration, by recrystallization in ethyl acetate were purified.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-phenylacetamide (12a)
Cream solid; Yield:65%;MP = 209–211 °C; IR (KBr, vmax) 3230 (NH), 3040 (CH Aromatic), 2930 (CH Aliphatic), 1666 (C = O) Cm− 1; 1H NMR (400 MHz, DMSO) δ 12.26 (s, 1 H), 11.65 (s, 1 H), 10.44 (s, 1 H), 8.54 (s, 1 H), 8.36 (s, 1 H), 8.01 (d, J = 8.3 Hz, 1 H), 7.84 (d, J = 8.3 Hz, 1 H), 7.73 (t, J = 7.6 Hz, 1 H), 7.62 (d, J = 8.7 Hz, 1 H), 7.52 (t, J = 7.6 Hz, 1 H), 7.34–7.32 (m, 1 H), 7.33–7.26 (m, 2 H), 7.13–7.09 (m, 3 H), 7.05 (d, J = 7.3 Hz, 2 H), 4.21 (s, 2 H), 3.34–3.31 (m, 2 H), 3.19 (t, J = 6.2 Hz, 2 H);13C NMR (101 MHz, DMSO) δ13C NMR (101 MHz, DMSO-d6) δ 173.95, 167.47, 157.05, 156.82, 154.95, 147.06, 142.09, 139.69, 139.32, 135.77, 131.43, 131.35, 129.25, 128.94, 127.54, 126.59, 126.26, 125.59, 123.69, 120.12, 119.54, 119.54, 35.93, 30.79, 23.76.; Anal. Calcd: C28H24N6O2S; C, 66.12; H, 4.76; N, 16.52; Found; C, 66.31; H, 4.92; N, 16.68.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazono)methyl)quinolin-2-yl)thio)-N-(m-tolyl)acetamide (12b)
Cream solid; Yield:78%;MP = 218–220 °C; IR (KBr, vmax) 3236 (NH), 3017 (CH Aromatic), 2948 (CH Aliphatic), 1666 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 12.25 (s, 1 H), 11.64 (s, 1 H), 10.35 (s, 1 H), 8.54 (s, 1 H), 8.36 (s, 1 H), 8.01 (d, J = 6.8 Hz, 1 H), 7.84 (d, J = 8.4 Hz, 1 H), 7.73 (t, J = 6.9 Hz, 1 H), 7.53 (t, J = 7.5 Hz, 1 H), 7.48 (s, 1 H), 7.39 (d, J = 8.2 Hz, 1 H), 7.18 (t, J = 7.8 Hz, 2 H), 7.14–7.09 (m, 2 H), 6.87 (d, J = 7.3 Hz, 1 H), 4.21 (s, 2 H), 3.36–3.30 (m, 2 H), 3.19 (t, J = 6.1 Hz, 3 H), 2.26 (s, 3 H)13C NMR (101 MHz, DMSO-d6) δ 173.95, 167.37, 156.82, 154.95, 147.07, 141.57, 139.67, 138.40, 135.74, 131.43, 131.35, 129.17, 129.09, 128.94, 127.56, 126.59, 126.27, 125.72, 125.59, 124.40, 120.06, 116.73, 35.94, 30.76, 23.75, 21.68; Anal. Calcd: C29H26N6O2S; C, 66.65; H, 5.01; N, 16.08; Found; C, 66.82; H, 5.23; N, 16.36.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(p-tolyl)acetamide (12c)
Cream solid; Yield:68%;MP = 196–198 °C; IR (KBr, vmax) 3250 (NH), 3010 (CH Aromatic), 2870 (CH Aliphatic), 1662 (C = O) Cm− 1;1 H NMR (400 MHz, DMSO) δ 12.26 (s, 1 H), 10.36 (s, 1 H), 9.07 (s, 1 H), 8.85 (s, 1 H), 8.06 (d, J = 7.9 Hz, 1 H), 7.90 (d, J = 8.2 Hz, 1 H), 7.83 (d, J = 6.7 Hz, 1 H), 7.59 (t, J = 7.6 Hz, 1 H), 7.53 (d, J = 8.8 Hz, 1 H), 7.52–7.49 (m, 3 H), 7.12 (d, J = 8.0 Hz, 2 H), 7.11–7.09 (m, 2 H), 4.24 (s, 2 H), 3.35–3.31 (m, 2 H), 3.23–3.16 (m, 2 H), 2.25 (s, 3 H);13C NMR (101 MHz, DMSO-d6) δ13C NMR (101 MHz, DMSO-d6) δ 173.54, 167.17, 159.51, 157.89, 149.68, 148.47, 147.85, 145.93, 141.68, 139.47, 137.24, 132.62, 130.53, 127.68, 126.82, 126.33, 125.50, 125.36, 120.17, 119.56, 119.53, 36.15, 30.76, 23.87 20.91; Anal. Calcd: C29H26N6O2S; C, 66.65; H, 5.01; N, 16.08; Found; C, 66.81; H, 5.23; N, 16.24.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(4-methoxyphenyl)acetamide (12d)
Cream solid; Yield:78%;MP = 223–225 °C; IR (KBr, vmax) 3236 (NH), 3017 (CH Aromatic), 2948 (CH Aliphatic), 1666 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 11.67 (s, 1 H), 10.29 (s, 1 H), 8.54 (s, 1 H), 8.37 (s, 1 H), 8.01 (d, J = 8.0 Hz, 1 H), 7.88 (d, J = 8.4 Hz, 1 H), 7.75 (t, J = 7.8 Hz, 1 H), 7.57–7.50 (m, 5 H), 7.21–7.16 (m, 2 H), 6.89 (d, J = 8.4 Hz, 4 H), 4.20 (s, 2 H), 3.73 (s, 3 H), 3.43–3.33 (m, 2 H), 3.28–3.26 (m, 2 H);13C NMR (101 MHz, DMSO-d6) δ 173.79, 166.93, 156.87, 155.71, 147.11, 141.71, 139.84, 137.87, 135.77, 134.26, 132.88, 131.40, 128.92, 127.58, 126.60, 126.25, 125.59, 122.35, 122.16, 121.15, 114.78, 114.38, 55.63, 35.83, 30.66, 23.50; Anal. Calcd: C29H26N6O3S; C, 64.67; H, 4.87; N, 15.60; Found; C, 64.82; H, 5.07; N, 15.76.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(2,3-dimethylphenyl)acetamide (12e)
Cream solid; Yield:63%;MP = 201–203 °C; IR (KBr, vmax) 3238 (NH), 3034 (CH Aromatic), 2942 (CH Aliphatic), 1663 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 12.25 (s, 1 H), 11.65 (s, 1 H), 9.78 (s, 1 H), 8.56 (s, 1 H), 8.38 (s, 1 H), 8.02 (d, J = 6.7 Hz, 1 H), 7.93 (d, J = 8.6 Hz, 1 H), 7.77 (t, J = 6.9 Hz, 1 H), 7.59–7.51 (m, 1 H), 7.48–7.47 (m, 1 H), 7.47–7.45 (m, 1 H), 7.13–7.12(m, 1 H), 7.12–7.11 (m, 1 H), 7.04–6.98 (m, 3 H), 4.25 (s, 2 H), 2.35–2.33 (m, 2 H), 3.20 (t, J = 6.0 Hz, 2 H), 2.21 (s, 3 H), 2.01 (s, 3 H).;13C NMR (101 MHz, DMSO) δ13C NMR (101 MHz, DMSO-d6) δ 173.94, 167.40, 156.84, 154.94, 147.16, 141.51, 139.65, 137.42, 136.60, 136.54, 135.67, 131.68, 131.35, 129.20, 128.97, 127.65, 127.42, 126.60, 126.45, 126.34, 125.65, 35.12, 30.75, 23.75, 20.57, 14.44.; Anal. Calcd: C30H28N6O2S; C, 67.14; H, 5.26; N, 15.66; Found; C, 67.32; H, 5.41; N, 15.89.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(2,4-dimethylphenyl)acetamide (12f)
Cream solid; Yield:63%;MP = 207–208 °C; IR (KBr, vmax) 3245 (NH), 3030 (CH Aromatic), 2930 (CH Aliphatic), 1664 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 11.68 (s, 1 H), 9.64 (s, 1 H), 8.57 (s, 1 H), 8.39 (s, 1 H), 8.02 (d, J = 6.8 Hz, 1 H), 7.96 (d, J = 8.2 Hz, 1 H), 7.77 (t, J = 7.7 Hz, 2 H), 7.53–7.51 (m, 2 H), 7.21–7.15 (m, 2 H), 7.04–7.03 (m, 2 H), 7.02–7.01 (m, 1 H), 4.30 (s, 2 H), 3.37 (t, J = 7.0 Hz, 1 H), 3.24 (t, J = 7.0 Hz, 1 H) 2.07 (s, 6 H) ;13C NMR (101 MHz, DMSO-d6) δ 173.76, 166.88, 156.74, 154.93, 147.22, 141.57, 139.78, 137.77, 135.73, 135.59, 135.52, 131.31, 128.96, 128.06, 127.70, 126.93, 126.61, 126.36, 125.81, 125.67, 122.35, 122.15, 114.71, 34.53, 30.62, 23.48, 18.49; Anal. Calcd: C29H26N6O2S; C, 66.65; H, 5.01; N, 16.08; Found; C, 66.81; H, 5.22; N, 16.25.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(2-chlorophenyl)acetamide (12 g)
Cream solid; Yield:71%;MP = 213–215 °C; IR (KBr, vmax) 3283 (NH), 3045 (CH Aromatic), 2962 (CH Aliphatic), 1666 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 11.66 (s, 1 H), 9.87 (s, 1 H), 8.56 (s, 1 H), 8.36 (s, 1 H), 8.02 (d, J = 7.2 Hz, 1 H), 7.93 (d, J = 8.4 Hz, 1 H), 7.81 (d, J = 6.6 Hz, 1 H), 7.75 (t, J = 7.7 Hz, 1 H), 7.55 (t, J = 7.5 Hz, 1 H), 7.49–748 (m, 1 H), 7.48–7.47 (m, 1 H), 7.45–7.44 (m, 1 H), 7.30 (t, J = 6.8 Hz, 1 H), 7.15–713 (m, 2 H), 7.11 (m, 1 H), 4.30 (s, 2 H), 3.35–3.34 (m, 2 H) 3.20 (t, J = 6.6 Hz, 4 H);13C NMR (101 MHz, DMSO-d6) δ 173.97, 168.07, 156.43, 154.96, 147.06, 141.59, 139.73, 136.20, 135.43, 131.37, 129.89, 128.91, 127.96, 127.70, 126.70, 126.38, 126.31, 125.79, 125.67, 125.39, 121.68, 121.60, 35.21, 30.77, 23.73; Anal. Calcd: C28H23ClN6O2S; C, 61.93; H, 4.27; N, 15.48; Found; C, 62.13; H, 4.46; N, 15.62.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(3-chlorophenyl)acetamide (12 h)
Cream solid; Yield:71%;MP = 219–221 °C; IR (KBr, vmax) 3279 (NH), 3039 (CH Aromatic), 2968 (CH Aliphatic), 1669 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 12.25 (s, 1 H), 11.65 (s, 1 H), 9.87 (s, 1 H), 8.57 (s, 1 H), 8.36 (s, 1 H), 8.02 (d, J = 6.8 Hz, 1 H), 7.93 (d, J = 8.4 Hz, 1 H), 7.81 (d, J = 8.1 Hz, 1 H), 7.76 (t, J = 7.6 Hz, 1 H), 7.55 (t, J = 7.5 Hz, 1 H), 7.49–7.43 (m, 2 H), 7.34–7.26 (m, 1 H), 7.17–7.15 (m, 1 H), 7.12–7.11 (m, 1 H), 7.11–7.09 (m, 1 H), 4.30 (s, 2 H), 3.36–3.32 (m, 2 H), 3.19 (t, J = 6.3 Hz, 3 H);13C NMR (101 MHz, DMSO) δ13C NMR (101 MHz, DMSO-d6) δ 173.98, 168.07, 156.44, 154.96, 147.07, 141.59, 139.72, 136.19, 135.43, 131.37, 129.89, 129.14, 128.91, 127.96, 127.70, 126.71, 126.38, 126.32, 125.80, 125.67, 125.39, 121.72, 121.64, 35.21, 30.78, 23.75; Anal. Calcd: C28H23ClN6O2S; C, 61.93; H, 4.27; N, 15.48; Found; C, 62.12; H, 4.42; N, 15.71.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(4-chlorophenyl)acetamide (12i)
Cream solid; Yield:71%;MP = 219–221 °C; IR (KBr, vmax) 3279 (NH), 3039 (CH Aromatic), 2968 (CH Aliphatic), 1669 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 12.25 (s, 1H), 11.64 (s, 1H), 10.59 (s, 1H), 8.54 (s, 1H), 8.35 (s, 1H), 8.00 (d, J = 8.2 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.72 (t, J = 7.0 Hz, 1H), 7.67–7.66 (m, 1H), 7.65–7.64 (m, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.40–7.38 (m, 2 H), 7.37–7.35 (m, 2 H), 7.15–7.06 (m, 2 H), 4.20 (s, 2 H), 3.35–3.33 (m, 2 H), 3.19 (t, J = 6.2 Hz, 2 H);13C NMR (101 MHz, DMSO-d6) δ 173.96, 167.72, 156.73, 154.96, 147.01, 143.68, 139.76, 138.72, 135.96, 131.38, 129.18, 128.92, 127.48, 127.19, 126.59, 126.23, 125.57, 124.46, 123.80, 121.85, 121.03, 118.55, 111.19, 35.99, 30.76, 23.74; Anal. Calcd: C28H23ClN6O2S; C, 61.93; H, 4.27; N, 15.48; Found; C, 62.11; H, 4.43; N, 15.71.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazono)methyl)quinolin-2-yl)thio)-N-(2,4-dichlorophenyl)acetamide (12j)
Cream solid; Yield:78%;MP = 205–207 °C; IR (KBr, vmax) 3310 (NH), 3020 (CH Aromatic), 2960 (CH Aliphatic), 1666 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 12.28 (s, 1 H), 11.65 (s, 1 H), 9.95 (s, 1 H), 8.56 (s, 1 H), 8.35 (s, 1 H), 8.02 (d, J = 7.3 Hz, 1 H), 7.90 (d, J = 8.4 Hz, 1 H), 7.82 (d, J = 8.9 Hz, 1 H), 7.75 (t, J = 7.7 Hz, 1 H), 7.64–7.63 (m, 1 H), 7.55 (t, J = 7.6 Hz, 1 H), 7.50–7.44 (m, 2 H), 7.40–7.37 (m, 1 H), 7.12–7.11 (m, 1 H), 7.11–7.10 (m, 1 H), 4.29 (s, 2 H), 3.35–3.32 (m, 2 H), 3.19 (t, J = 6.8 Hz, 2 H);13C NMR (101 MHz, DMSO-d6) δ 173.96, 168.26, 156.41, 154.95, 147.03, 141.61, 140.67, 139.78, 136.30, 134.65, 131.45, 131.39, 129.33, 128.91, 128.09, 127.66, 126.72, 126.38, 126.30, 125.66, 121.68, 121.61, 117.93, 35.23, 32.14, 30.77, 23.72; Anal. Calcd: C28H22Cl2N6O2S; C, 58.24; H, 3.84; N, 14.55; Found; C, 58.41; H, 4.11; N, 14.69.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(4-nitrophenyl)acetamide (12k)
Cream solid; Yield:78%;MP = 224–226 °C; IR (KBr, vmax) 3320 (NH), 3056 (CH Aromatic), 2962 (CH Aliphatic), 1671 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 11.83–11.27 (m, 1 H), 11.66 (s, 1 H), 11.12 (s, 1 H), 9.07 (s, 1 H), 8.85 (s, 1 H), 8.28–8.26 (m, 1 H), 8.25–8.23 (m, 2 H), 8.26–8.19 (m, 1 H), 8.09–8.07 (m, 1 H), 7.92 (d, J = 9.2 Hz, 2 H), 7.89 (d, J = 9.1 Hz, 1 H), 7.80–7.68 (m, 1 H), 7.75 (d, J = 8.4 Hz, 1 H), 7.58 (t, J = 6.0 Hz, 1 H), 7.50 (d, J = 7.3 Hz, 1 H), 7.18–7.11 (m, 1 H), 4.30 (s, 2 H), 3.26–3.15 (m, 2 H);13C NMR (101 MHz, DMSO) δ13C NMR (101 MHz, DMSO-d6) δ 175.93, 168.76, 159.66, 157.63, 147.72, 145.94, 142.63, 139.98, 137.14, 135.41, 131.60, 129.40, 128.36, 127.52, 126.89, 126.65, 125.62, 125.47, 123.58, 121.91, 119.16, 36.48, 30.71, 23.60.Anal. Calcd: C28H23N7O4S; C, 60.75; H, 4.19; N, 17.71; Found; C, 60.92; H, 4.37; N, 17.94.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(2-methyl-4-nitrophenyl)acetamide (12 L)
Cream solid; Yield:61%;MP = 238–240 °C; IR (KBr, vmax) 3330 (NH), 3060 (CH Aromatic), 2967 (CH Aliphatic), 1672 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 11.67 (s, 1 H), 9.98 (s, 1 H), 8.57 (s, 1 H), 8.36 (s, 1 H), 8.15–8.13 (m, 1 H), 8.07–8.04 (s, 1 H), 8.02 (d, J = 8.3 Hz, 1 H), 7.93–7.91 (m, 1 H), 7.83 (d, J = 8.4 Hz, 1 H), 7.74 (t, J = 8.4 Hz, 1 H), 7.58–7.50 (m, 1 H), 7.50–7.47 (m, 2 H), 7.14–7.13 (m, 21 H), 7.12–7.11 (m, 1 H), 4.34 (s, 2 H), 3.21 (t, J = 7.4 Hz, 2 H), 2.33 (s, 3 H) ;13C NMR (101 MHz, DMSO) δ13C NMR (101 MHz, DMSO-d6) δ 173.92, 168.31, 156.57, 154.96, 147.28, 147.01, 143.59, 143.47, 141.64, 139.85, 136.25, 131.46, 128.97, 127.49, 126.69, 126.42, 126.29, 125.95, 125.65, 123.47, 122.32, 121.80, 119.55, 118.73, 35.58, 30.73, 23.64, 18.26.; Anal. Calcd: C29H25N7O4S; C, 61.36; H, 4.44; N, 17.27; Found; C, 61.53; H, 4.67; N, 17.49.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(2-fluorophenyl)acetamide (12 m)
Cream solid; Yield:64%;MP = 227–229 °C; IR (KBr, vmax) 3285 (NH), 3035 (CH Aromatic), 2960 (CH Aliphatic), 1670 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 11.65 (s, 1 H), 10.22 (s, 1 H), 8.56 (s, 1 H), 8.35 (s, 1 H), 8.02 (d, J = 8.1 Hz, 1 H), 7.90 (d, J = 8.2 Hz, 1 H), 7.76 (t, J = 7.7 Hz, 1 H), 7.54 (t, J = 7.6 Hz, 1 H), 7.50–7.49 (m, 1 H), 7.49–7.47 (m, 1 H), 7.30–7.23 (m, 2 H), 7.15–7.13 (m, 2 H), 7.13–7.11 (m, 2 H), 4.27 (s, 2 H), 3.36–3.33 (m, 2 H), 3.20 (t, J = 7.2 Hz, 2 H);13C NMR (101 MHz, DMSO) δ13C NMR (101 MHz, DMSO-d6) δ 173.92, 168.12, 156.71, 155.03, 154.95, 152.58, 147.49, 147.06, 139.76, 136.08, 131.37, 129.12, 128.90, 127.52, 126.83, 126.66, 126.26, 125.62, 124.86, 124.13, 121.76, 116.02, 115.84, 35.39, 30.72, 23.66; Anal. Calcd: C28H23FN6O2S; C, 63.86; H, 4.40; N, 15.96; Found; C, 64.05; H, 4.56; N, 16.13.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-(4-fluorophenyl)acetamide (12n)
Cream solid; Yield:61%;MP = 230–232 °C; IR (KBr, vmax) 3283 (NH), 3041 (CH Aromatic), 2975 (CH Aliphatic), 1673 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 12.25 (s, 1 H), 11.64 (s, 1 H), 10.49 (s, 1 H), 8.54 (s, 1 H), 8.36 (s, 1 H), 8.00 (d, J = 9.6 Hz, 1 H), 7.83 (d, J = 8.4 Hz, 2 H), 7.75–7.72 (m, 1 H), 7.66–7.64 (m, 1 H), 7.64–7.63 (m, 1 H), 7.63–7.61 (m, 1 H), 7.55–7.50 (m, 2 H), 7.18–7.16 (m, 1 H), 7.15–7.14 (m, 1 H), 7.13–7.11 (m, 2 H), 4.20 (s, 2 H), 3.37–3.31 (m, 2 H) 3.19 (t, J = 6.2 Hz, 3 H);13C NMR (101 MHz, DMSO-d6) δ 173.96, 167.43, 159.62, 157.23, 156.77, 154.96, 147.04, 141.61, 139.73, 136.15, 135.86, 131.37, 129.15, 128.93, 127.51, 126.59, 126.35, 126.24, 125.58, 121.31, 121.24, 115.93, 115.71, 111.24, 35.88, 30.77, 23.75; Anal. Calcd: C28H23FN6O2S; C, 63.86; H, 4.40; N, 15.96; Found; C, 64.08; H, 4.56; N, 16.10.
(E)-2-((3-((2-(3-(1 H-benzo[d]imidazol-2-yl)propanoyl)hydrazineylidene)methyl)quinolin-2-yl)thio)-N-benzylacetamide (12o)
Cream solid; Yield:71%;MP = 227–229 °C; IR (KBr, vmax) 3260 (NH), 3045 (CH Aromatic), 2962 (CH Aliphatic), 1662 (C = O) Cm− 1;1H NMR (400 MHz, DMSO) δ 11.69 (s, 1 H), 10.54–10.45 (m, 1 H), 8.53 (s, 1 H), 8.36 (s, 1 H), 7.99 (d, J = 6.6 Hz, 1 H), 7.84 (d, J = 8.4 Hz, 1 H), 7.73 (t, J = 6.9 Hz, 2 H), 7.59–7.58 (m, 1 H), 7.57–7.58 (m, 1 H), 7.53 (t, J = 7.0 Hz, 2 H), 7.26–7.25 (m, 1 H), 7.24–7.23 (m, 1 H), 7.18–7.13 (m, 3 H), 4.25 (d, J = 7.6 Hz, 1 H) 4.21 (s, 2 H), 3.41 (t, J = 6.9 Hz, 2 H), 3.29 (t, J = 7.0 Hz, 2 H);13C NMR (101 MHz, DMSO-d6) δ 173.65, 167.43, 156.76, 154.98, 147.05, 140.05, 136.15, 136.05, 131.42, 128.90, 127.53, 126.62, 126.17, 125.54, 123.04, 122.65, 121.32, 121.24, 115.93, 115.71, 114.63, 42.15, 35.92, 30.54, 23.21.Anal. Calcd: C29H26N6O2S; C, 66.65; H, 5.01; N, 16.08; Found; C, 66.82; H, 5.21; N, 16.22.
α-Glucosidase and α-amylase inhibitory activity assay
The inhibitory activities of the new compounds 12a-o against α-glucosidase and α-amylase were assessed by the reported protocols by Tao et al. and Xiao et al., respectively31,32. For the α-glucosidase assay, the used substrate p-nitrophenyl-α-D-glucopyranoside (p-NPG) dissolved in a phosphate buffer solution at pH 7.4 and a concentration of 5 mM. To prepare sample dilutions, 5 mg of the synthesized compounds were dissolved in 5 mL of a mixture of ethanol and water. Various dilutions of the samples in phosphate buffer were then prepared to achieve complete enzyme inhibition. For the α-amylase assay, a starch substrate solution was prepared by dissolving 3 g of starch in 150 mL of 0.4 M NaOH and heating it at 98 °C for 10 min. After cooling the solution and adjusting the pH to 7.0 with 2.0 M HCl, the volume was brought up to 200 mL with water. Next, a mixture consisting of 200 µL of phosphate buffer, 50 µL of the substrate, and 5-200 µL of the sample solution was pre-incubated at 35 °C for 3 min for the α-glucosidase assay and 30 min for the α-amylase assay. The absorbance readings for the α-glucosidase and α-amylase inhibition assays were measured at 405 nm and 580 nm, respectively. These readings were used to determine the inhibitory activities of the synthesized compounds33.
Molecular Docking study
The synthesized compounds underwent molecular docking investigations using the Smina molecular docking program34. Docking studies of the most potent compound 12n was performed on α-glucosidase and α-amylase based on our previously reported work35. The X-ray crystal structures of α-glucosidase (PDB code: 5NN8) and α-amylase (PDB code: 4w93) were obtained from the Protein Data Bank (http://www.rcsb.org). Native ligands and water molecules were removed, and the protein structures were repaired before the docking procedure. Non-polar hydrogen atoms were merged, and polar hydrogen atoms were added. The protein’s Kollmann charges were computed. The compound structures were drawn using MarvinSketch version 20.11.0 (ChemAxon, Budapest, Hungary, http://www.chemaxon.com) and then optimized using the semi-empirical AM1 method through the SCBDD server (https://www.scbdd.com/mopac-optimization/optimize/). Open Babel 2.4.1 was used to assign the ligands their Gasteiger partial charges. The co-crystalline ligand’s coordinates were used to automatically define the binding site. For α-glucosidase, the grid box was centered at x = − 13.872 Å, y = − 37.459 Å, z = 95.384 Å, with dimensions of 40 × 58 × 40 points and a grid spacing of 0.375 Å. For α-amylase, the grid box was centered at x = − 10.922 Å, y = 4.087 Å, z = − 25.289 Å, with dimensions of 66 × 56 × 55 points and the same grid spacing. Discovery Studio 2019 molecular visualization software (https://discover.3ds.com/) was utilized to visualize the interactions between the proteins and ligands.
Molecular dynamics simulation studies
To further analyze the behavior and bond stability of the potent inhibitor 12n in the active sites of α-glucosidase and α-amylase, molecular dynamics simulations were performed36. These simulations were conducted on a 24-core Linux GPU server using the GROMACS 4.6.5 simulation package. The Amber99sb force field was employed to determine atom types and simulation parameters, with a temperature of 300 K and a pH of 7.4. Topology and coordinate files for the inhibitors were generated using the acpype program. To create the simulation environment, a rhombic dodecahedral box was defined and filled with the TIP3P water model, and neutralization was achieved by adding sodium chloride ions. The entire system underwent equilibration under the NVT ensemble for 500 ps, followed by an additional equilibration phase under the NPT ensemble for 500 ps, during which the system’s pressure stabilized at 1 atm. The molecular dynamics run was carried out with a sampling time of 2 fs, spanning a total time of 100 ns. Towards the end of the simulation, the periodic boundary conditions of the trajectory were removed, and the protein was repositioned to the center of the box37. Indicators of structural stability, such as the root-mean-square deviation (RMSD) in Angstroms (Å), root-mean-square fluctuation (RMSF) values for the protein’s backbone atoms, radius of gyration (Rg), and the total number of hydrogen bonds (H-bonds) between the protein and ligands during the simulation time, were calculated. Finally, the trajectory was clustered during the equilibrium time range using the Gromos method with a cut-off value of 0.25. Discovery Studio 2019 molecular visualization software was employed to visualize the interactions.
In silico pharmacokinetics
Druglikeness and ADMET properties of the most potent compounds were predicted by PreADMET, SwissADME, and ADMETlab 2.0 as three credible online servers38,39,40.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Lin, X. et al. Global, regional, and National burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. Sci. Rep. 10, 1–11 (2020).
Toender, A., Vestergaard, M., Munk-Olsen, T. & Laursen, L. J. T. K. J. K. Risk of diabetic complications and subsequent mortality among individuals with schizophrenia and diabetes-a population-based register study. Schizophr Res. 218, 99–106 (2020).
Kumawat, V. S. & Kaur, G. Therapeutic potential of cannabinoid receptor 2 in the treatment of diabetes mellitus and its complications. Eur. J. Pharmacol. 862, 172628 (2019).
Dewi, E. U. et al. The relationship between diabetes self-care management and blood glucose level among type 2 diabetes mellitus patients. Int. J. Public. Heal Sci. 12, 1165 (2023).
Asif, M. The prevention and control the type-2 diabetes by changing lifestyle and dietary pattern. J. Educ. Health Promot. 3, 1 (2014).
Janeček, Š., Svensson, B. & MacGregor, E. A. α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell. Mol. Life Sci. 71, 1149–1170 (2014).
Williams, J. A. & Amylase Pancreapedia: The Exocrine Pancreas Knowledge Base (2019).
Adisakwattana, S. Cinnamic acid and its derivatives: mechanisms for prevention and management of diabetes and its complications. Nutrients 9, 163 (2017).
Okuyama, M., Saburi, W., Mori, H. & Kimura, A. α-Glucosidases and α-1, 4-glucan lyases: structures, functions, and physiological actions. Cell. Mol. Life Sci. 73, 2727–2751 (2016).
Derosa, G. & Maffioli, P. α-Glucosidase inhibitors and their use in clinical practice. Arch. Med. Sci. 8, 899 (2012).
Jakhar, R., Dangi, M., Khichi, A. & Chhillar, A. K. Relevance of molecular Docking studies in drug designing. Curr. Bioinform. 15, 270–278 (2020).
Brogi, S., Ramalho, T. C., Kuca, K., Medina-Franco, J. L. & Valko, M. In Silico methods for drug design and discovery. Front. Chem. 8, 612 (2020).
Rao, V. S. & Srinivas, K. Modern drug discovery process: an in Silico approach. J. Bioinform Seq. Anal. 2, 89–94 (2011).
Yang, S-Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today. 15, 444–450 (2010).
Ferreira, L. G., Dos Santos, R. N., Oliva, G. & Andricopulo, A. D. Molecular Docking and structure-based drug design strategies. Molecules 20, 13384–13421 (2015).
Cetiz, M. V. et al. Bioinformatic and experimental approaches to uncover the bio-potential of mercurialis annua extracts based on chemical constituents. J. Mol. Liq. 427, 127390 (2025).
Zheleva-Dimitrova, D. et al. Exploring hidden natural resources for bioactive compounds: focused chemical and biological studies on some anthemis species. Food Chem. Toxicol 115467 (2025).
Llorent-Martinez, E. J. et al. Characterization of the chemical profiles and biological activities of thesium Bertramii azn. Extracts using a combination of in vitro, in silico, and network Pharmacology methods. Fitoterapia 180, 106329 (2025).
Matada, B. S., Pattanashettar, R. & Yernale, N. G. A comprehensive review on the biological interest of Quinoline and its derivatives. Bioorg. Med. Chem. 32, 115973 (2021).
Rahim, F. et al. Triazinoindole analogs as potent inhibitors of α-glucosidase: synthesis, biological evaluation and molecular Docking studies. Bioorg. Chem. 58, 81–87 (2015).
Karami, M. et al. One-pot multi-component synthesis of novel Chromeno [4, 3-b] pyrrol-3-yl derivatives as alpha-glucosidase inhibitors. Mol. Divers. 25, 1–3 (2012).
Zarenezhad, E., Farjam, M. & Iraji, A. Synthesis and biological activity of pyrimidines-containing hybrids: focusing on Pharmacological application. J. Mol. Struct. 1230, 129833 (2021).
Safapoor, S. et al. Synthesis, ADMT prediction, and in vitro and in Silico a-glucosidase Inhibition evaluations of new quinoline–quinazolinone–thioacetamides. RSC Adv. 13, 19243–19256 (2023).
Forozan, R. et al. Synthesis, in vitro inhibitor screening, structure–activity relationship, and molecular dynamic simulation studies of novel thioquinoline derivatives as potent α-glucosidase inhibitors. Sci. Rep. 13, 7819 (2023).
Noori, M. et al. Design, synthesis, and in Silico studies of quinoline-based-benzo [d] imidazole bearing different acetamide derivatives as potent α-glucosidase inhibitors. Sci. Rep. 12, 14019 (2022).
Aroua, L. M. et al. A facile approach synthesis of benzoylaryl benzimidazole as potential α-amylase and α-glucosidase inhibitor with antioxidant activity. Bioorg. Chem. 114, 105073 (2021).
Khan, S. et al. Synthesis, in vitro α-amylase, α-glucosidase activities and molecular Docking study of new benzimidazole bearing Thiazolidinone derivatives. J. Mol. Struct. 1269, 133812 (2022).
Hussain, R. et al. Synthesis of novel benzimidazole-based thiazole derivatives as multipotent inhibitors of α-amylase and α-glucosidase: in vitro evaluation along with molecular Docking study. Molecules 27, 6457 (2022).
Mohammadizadeh, S. et al. Design of novel benzimidazole-propane Hydrazide derivatives as α-glucosidase and α-amylase inhibitors: in vitro and in Silico studies. Med. Chem. Res. 34, 205–218 (2025).
Abbasi, I. et al. Isatin-hydrazide conjugates as potent α-amylase and α-glucosidase inhibitors: synthesis, structure and in vitro evaluations. Bioorg. Chem. 116, 105385 (2021).
Tao, Y., Zhang, Y., Cheng, Y. & Wang, Y. Rapid screening and identification of α-glucosidase inhibitors from mulberry leaves using enzyme‐immobilized magnetic beads coupled with HPLC/MS and NMR. Biomed. Chromatogr. 27, 148–155 (2013).
Xiao, Z., Storms, R. & Tsang, A. A quantitative starch? Iodine method for measuring alpha-amylase and glucoamylase activities. Anal. Biochem. 351, 146–148 (2006).
Taslimi, P., Akıncıoglu, H. & Gülçin, İ. Synephrine and phenylephrine act as α-amylase, α‐glycosidase, acetylcholinesterase, butyrylcholinesterase, and carbonic anhydrase enzymes inhibitors. J. Biochem. Mol. Toxicol. 31, e21973 (2017).
Koes, D. R., Baumgartner, M. P. & Camacho, C. J. Lessons learned in empirical scoring with Smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 53, 1893–1904 (2013).
Karimian, S. et al. Synthesis and biological evaluation of benzimidazoles/1, 3, 5-triazine-2, 4-diamine hybrid compounds: a new class of multifunctional alzheimer targeting agents. New. J. Chem. 46, 15567–15584 (2022).
Ghanbarlou, M. et al. 4-Phenylthiazol-1, 2, 3-triazole derivatives as new potential α-glucosidase and α-amylase inhibitors. J. Mol. Struct. 1334, 141919 (2025).
Naimi, N. et al. New benzimidazole–indole–amide derivatives as potent α-glucosidase and acetylcholinesterase inhibitors. Arch Pharm 358, e2400354 .
Bioinformatics and Molecular Design Research Center, PreADMET program. Bioinformatics and Molecular Design Research Center, Seoul, South Korea. (2014). http://preadmet.bmdrc.org
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Xiong, G. et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 49, W5–14 (2021).
Author information
Authors and Affiliations
Contributions
P.N., S.S., M.N., N.D., and S.N.G. contributed in the synthesis and characterization of compounds. P.T. performed in vitro biological assay. S.K. performed in silico assays. B.L., M.M-K., and M.M. conceived the idea and designed the experiments. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nikfar, P., Karimian, S., Safapoor, S. et al. Introduction of new quinolone-2-thio-acetamide-propane hydrazide-benzimidazole derivatives as new α-glucosidase and α-amylase inhibitors. Sci Rep 15, 31349 (2025). https://doi.org/10.1038/s41598-025-16661-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-16661-7
