
Abstract
Serenoa repens (Bartram) J.K.Small known as Saw palmetto; its herb is widely used for its therapeutic potentials especially in the treatment of urological disorders such as benign prostatic hyperplasia (BPH) in men. Comparative chemical, biological and in silico docking profiling’s of Serenoa repens seeds extract (S1) vs. a particular pharmaceutical product (S2) containing Serenoa repens. LCHR-ESI-MS is used for identification of bioactive constituents. MTT Cytotoxic study using Pc3 and DU-145 prostate cancer cell lines is used to measure the cytotoxicity of S1 and S2. Molecular Dynamic (MD) Simulation studies were conducted on the top-scoring docked compounds using Desmond in the Schrödinger software package. The chemical analysis by LCHR-ESI-MS detected about fifty bioactive constituents of various classes in both samples under investigation mainly polyphenols like flavonoids, phenolic acids and some phytosterols. MTT Cytotoxic study using Pc3 and DU-145 prostate cancer cell lines revealed the pharmacological actions, such as anti-inflammatory, pro-apoptotic and anti-androgenic potentials of S1 and S2 against Pc3; (20.04 ± 0.89 and 7.30 ± 0.32), respectively and against DU-145; (50.74 ± 3.06 and 36.64 ± 1.97), respectively. A focused investigation of the plant extracts’ most prevalent and possibly bioactive substances which was evaluated by molecular docking and Molecular Dynamic (MD) Simulation studies, indicated that Quercetin-3-(6’’-malonyl)-Glucoside followed by Luteolin 7-(6’’-malonylneohesperidoside) have substantial inhibitory potential against CDK2. These results highlight Serenoa repens’ potential as a natural cure and direct the development of pharmaceutical formulations for improved therapeutic efficacy.
Similar content being viewed by others

Introduction
BPH (Benign prostatic hyperplasia) and prostate cancer are two most common urinary diseases affecting aging men1Prostate gland noncancerous growth (Benign prostatic hyperplasia; BPH) has resulted from overproliferation of the glandular and stromal tissue of the prostate accompanied by lower urinary tract symptoms which in some cases may lead to prostate cancer2. The rates of incidence of both are currently indicate a continued increase. Age is one of the most significant risk factors in the incidence of both illnesses3. Other specific risk factors are PV (prostate volume), PSA (serum prostate-specific antigen) and LUTS. BPH is characterized by proliferation of epithelial cells of the prostate and can lead to LUTS (lower urinary tract symptoms). Lower urinary tract symptoms together with an enlarged prostate form a multifaceted set of signs lead to direct BOO (bladder outlet obstruction)4. BPH affects approximately 30% of men over the age of 65. Nocturia and changes in urinary flow stream are the most predictive symptoms4.
For instance, men with prostate volume ≥ 30 mL and PSA ≥ 1.5 ng/mL have a significantly increased risk of BPH progression5. Herbal medicines are those that are made from plant materials and used to treat and improve human health6. Numerous herbal remedies were prescribed for relief of BPH symptoms as Serenoa repens fruits and pumplin seed oil that contain free fatty acids ad phytosterols7. Herbal medicines must receive the same level of quality control as pharmaceuticals made using chemicals. But regrettably, compared to synthetic pharmaceuticals, the regulations for herbal medicines are less stringent8. This is causing the quality standards of herbal products to decline through deliberate and sometimes unintentional adulteration, fake drugs, drug substitution, and many other methods that are likely to lower the quality of the herbal materials that are marketed and consumed for a healthy existence8. However, this approach has hazardous consequences for the health of consumers. Controlling the quality standards of herbal medicines and goods is therefore crucial for the welfare of humanity9.
The palm family Arecaceae, which contains evergreen trees, shrubs, and occasionally vines, includes the saw palmetto as a member. Saw palmetto is frequently found in the southern U.S. and the West Indies. A portion of the vegetation of the Florida Everglades is made up of saw palmettos10. They develop into enormous mounds of leaves when left alone. Recently, the saw palmetto has attracted interest as a potential prostate cancer treatment11. Serenoa repens W. Bartram (Saw palmetto), Figure S1, was selected based on the prevalence of its particular significance in men power activities beside the few reports on its chemical composition and biological effects12. Due to the high chemical and biological diversity of their metabolites, plants pose a significant challenge in metabolomics. The diverse chemical makeup of Serenoa repens is one of its unique traits. Many different bioactive substances, such free and esterified fatty acids, phosphoglycerides, flavonoids, tannins, terpenes, and other secondary metabolites, are found in it13. Indigenous societies have used these plants for ages in significant amounts as folk medicine to increase men’s potency and its oil as hair tonic14. Various pharmacological actions, such as anti-inflammatory and pro-apoptotic effects and anti-androgenic potentials15. but the precise bioactive constituents and molecular mechanisms underlying Serenoa repens’ effects in prostatic disease remain poorly understood. We aimed to address this gap by investigating Serenoa repens extract versus a formulated product, focusing on their anti-proliferative effects and potential CDK2 inhibition. Notably, persistent BPH and associated chronic inflammation can create a microenvironment permissive to prostate carcinoma in aging men5. Furthermore, CDK2/cyclin E activity is implicated in unchecked cell cycle progression in cancer16providing rationale for exploring Serenoa’s phytochemicals as potential CDK2 inhibitors. To our knowledge, no prior studies have examined Serenoa repens in this context. It is noteworthy that Serenoa repens extracts can induce cell-cycle arrest and apoptosis in cancer cells (e.g., by upregulating the cyclin-dependent kinase inhibitor p27 and inhibiting STAT3 signaling17, which indirectly supports our focus on CDK2. As a result, there is not a single analytical technique that can identify all plant metabolites at once. Both LCHR-ESI-MS and molecular docking are considere as powerfull strategies for plant profiling. It is thought that liquid chromatography-mass spectrometry, such as LCHR-ESI-MS, is an effective analytical method for metabolic profiling since it can identify a variety of chemical compounds without the need for time-consuming isolation procedures18. Molecular docking serves as a preliminary screening tool to identify natural compounds with potential CDK2 interactions by assessing binding affinities and modes18. Molecular dynamics (MD) simulations further validate these interactions by analyzing ligand-protein complexes over time, offering a dynamic perspective on stability and allosteric modulation. ADMET profiling evaluates the pharmacokinetic properties of lead compounds to predict their drug suitability, focusing on bioavailability, metabolism, and toxicity.
In conclusion, Serenoa repens was the plant we chose for this study in order to learn more about its metabolites and biological potentials. This decision placed our project in a position where it may be possible to find novel therapeutic agents and advance environmentally friendly practices through the analysis of their chemical constituents and pharmacological actions. Together, these computational methods form the backbone of our approach to identifying and characterizing novel natural products as selective CDK2 inhibitors. By combining these techniques, the study aims to establish a well-rounded understanding of each candidate’s potential, setting the stage for future empirical validations.
Experimental
Plant materials
Five hundred grams of Saw palmetto seeds were purchased from authenticated HARAZ herbal stores, Egypt, checked in the Faculty of Agriculture’s Taxonomy Department at Fayoum University, and kept in the Department of Pharmacognosy, Faculty of Pharmacy, Fayoum University, with a voucher sample ID (No. FuPD-5), and powdered. A capsule product of Saw palmetto was also bought from a pharmaceutical market and its contents were emptied. Both seeds and product and extracted by cold maceration with 70% ethanol three successive times, then filtered and concentrated by rotatory evaporator to give dried crude extracts S1 (seeds) and S2 (capsules), which used for comparative analytical and biological investigations.
LC-HR-ESI-MS
The analysis of the extract (1 mg/mL in MeOH) was conducted using an Accela HPLC system coupled with an Exactive (Orbitrap) mass spectrometer (Thermo Fisher Scientific) The compounds were separated on a Zorbax RP-18 column from Agilent Technologies (dimensions:150 mm × 3 mm, dp = 2.7 μm), Column temperature 35.0 °C, flow rate 0.3 ml/min. According to reported method19. The injection volume was 5 µL and a gradient elution method was employed, involving a mobile phase composed of 0.1% formic acid in water (A) and acetonitrile (B). Beginning at a flow rate of 300 µL/min with 10% B, the gradient elution linearly climbed to 100% B in 30 min, stayed isocratic for the next five minutes, and then linearly decreased back to 10% B for the next minute. Before the subsequent injection, the mobile phase was allowed to equilibrate for nine minutes. Mass spectrometry data (m/z 100–2000) was obtained using in-source CID, with MS/MS analysis for the m/z range 50–1000. MZmine 2.12 software facilitated differential analysis through chromatogram deconvolution, peak alignment, and gap-filling. The metabolites were identified by dereplicating each m/z ion peak against a customized database, and the putative identities of the top 20 features were determined by peak intensity. In-house and online databases were utilized to verify the identities of the detected metabolites.
Cytotoxic assay
Cell lines are gathered from VACSERA, Giza, Egypt, originally ATTC. MTT tetrazolium assay technology is a precise method gives accurate results, it was the first homogenous cell viability test created for high throughput screening (HTS) in a 96-well format which for this reason became popular and widespread through thousands of academic publications20. According to reported method21; Each well received 25 µL of a stock MTT at a concentration of 5 mg/ml in PBS solution and 100 µL of the samples concentrations (0.4, 1.6, 6.3, 25 and 100 µg), which was then incubated for 4 h. After aspirating the wells, 100 µL of 0.04 N HCI in isopropanol was used to lyse the cells and a flow Muhiskan MCC plate reader was used to read the plate at 570 nm. 3-[4,5-dimethylthiazol-2-yl]−2,5-diphenyl tetrazolium bromide is the key ingredient in MTT and the yielding purple solution is spectrophotometrically measured and subsequently the amount of formazan produced concomitant changes with changes in the number of cells, showing the level of cytotoxicity the test material. The assay is performed on the two samples; S1 and S2 and also staurosporine (positive control), all assays performed against blank negative control with complete medium without cells.
In-silico docking and molecular simulation
This study leveraged the well-documented properties of sunitinib and staurosporine as benchmark inhibitors to validate the efficacy of several natural products. By drawing comparisons with these known inhibitors, the aim was to explore the structural and binding attributes of these natural compounds as potential selective kinase inhibitors, aiming to achieve enhanced therapeutic outcomes with fewer side effects22.
Protein and ligands preparation
The X-ray crystal structure of Cyclin-dependent kinase 2 (CDK2) in complex with sunitinib (PDB ID: 3TI1) was sourced from the RCSB Protein Data Bank23. Protein modeling was performed using Schrödinger’s Protein Preparation Wizard24. Ligands, including the control inhibitors (sunitinib and staurosporine) and ten saw Palmetto compounds, were prepared using Schrödinger’s LigPrep (LigPrep 2.5, Schrödinger, LLC, New York, 2024), which optimized geometries and generated different bioactive forms. Prepared ligands were then docked into the CDK2 binding grid. Two known CDK inhibitors, sunitinib and staurosporine, were also docked into the CDK2 active site as positive controls to benchmark the docking protocol.
Grid generation and molecular docking
The binding site of CDK2 was defined using the ligand coordinates of the co-crystallized structure. Ligands were docked using the Glide XP docking protocol with specific settings to refine the docking process. The method involved the calculation of key scores (G score, Emodel, and XP GScore) to evaluate the binding efficiency. MM-GBSA was employed to rescore the docked poses, providing estimates for the binding free energies. The docking grid box was centered at the CDK2 ATP-binding site (based on the co-crystal ligand) with an inner box of ~ 20 × 20 × 20 Å and an outer box of 40 Å, encompassing all key active-site residues25. No constraints were applied during docking (i.e., no forced H-bonds), and XP mode was used for higher accuracy. To validate the protocol, the co-crystallized inhibitor sunitinib was re-docked into CDK2, yielding an RMSD of 0.60 Å from the crystal pose, confirming the docking method’s reliability.
Induced fit docking
IFD was used to account for receptor flexibility by generating multiple poses for the CDK2 receptor in response to ligand binding using the IFD protocol in Schrödinger Maestro v14.1 (Induced Fit Docking protocol; Glide, Schrödinger, LLC, New York, NY, 2024; Prime, Schrödinger, LLC, New York, NY, 2024). Glide docking was initially applied, followed by energy minimization of residues near the binding site. This approach allowed for refined predictions of receptor-ligand interactions.
Molecular dynamic (MD) simulation
MD simulations were conducted on the top-scoring docked compounds using Desmond in the Schrödinger software package (Desmond, Schrödinger, LLC, New York, 2024; D. E. Shaw J. Comput. Chem., 2005, 26, 1318 —1328), to provide insights into the dynamic stability of the CDK2-ligand complexes. The system was solvated in a water box and neutralized, and simulations were run for 200 ns under constant 1 atm pressure at 300 K. Simulations employed the OPLS4 force field (Schrödinger software suite versions 2024). Prior to production, each protein–ligand system was energy-minimized (up to 5,000 steps, steepest descent) and equilibrated for 1 ns under NPT conditions (300 K, 1 atm). Production MD runs (200 ns per complex) used a 2 fs time step. Each system was solvated in an orthorhombic water box with a 10 Å buffer and neutralized with counter-ions. Trajectory analyses (RMSD, hydrogen-bond occupancy, residue interaction frequency) were performed using Schrödinger’s Simulation Interaction Diagram tools and cross-checked with VMD.
Quantum mechanics/molecular mechanics (QM/MM) calculations
To delve deeper into binding interactions, QM/MM calculations were conducted, with the QM region (key CDK2 residues and ligands) using the Jaguar package, integrated within the Maestro suite (Jaguar, Schrödinger, LLC, New York, 2024), modeled at the B3LYP/6-31G* level. This method provided detailed insights into the electronic properties governing the binding interactions26.
Prediction of ADMET properties
ADMET properties of the CDK2 inhibitors were predicted using QikProp (QikProp, Schrödinger, LLC, New York, NY, 2024), to assess bioavailability and toxicity. This provided an additional layer of evaluation to prioritize the most promising compounds before experimental validation, ensuring a robust understanding of their potential as anticancer agents.
By summarizing detailed settings such as QM/MM and focusing on key steps and methodologies, this overview highlights the critical aspects of the in-silico process, facilitating an understanding of the efficacy and potential of these natural compounds as selective CDK2 inhibitors.
Statistical analysis
-IC50 values were calculated by Sigma Plot software, V.12(CA, USA) from the concentration- response curve by use of this equation:
% Cell viability = (100 - R) × [1 - [D]m /Kd m + [D]m] + R.
R: resistance fraction, D: used drug concentration used, m: a Hill-type coefficient, Kd: drug concentration producing a 50% reduction of the max. inhibition rate.
-One-way ANOVA was used to analyse the biochemical data, which were expressed as means ± SE. Duncan’s multiple range tests and least-significant difference test were used to compare the means between groups. The statistical significance was determined by a P-value < 0.05 (Graph Pad Prism 5).
Results and discussion
LC-HR-ESI-MS
Chemical LC-MS analysis reveals substantial variations in compounds concentrations between both plants extract (S1) and product (S2) which might be due to extraction methods or stability (Table 1), and The chromatograms of LCHR-ESI-MS (Figure S2), is shown in supplentary data.
As shown in Table 1, some compounds retained their concentrations in both samples as Amorphigenin and Astragalin, while some components are present in higher concentration in product more that the plant samples as Epicatechin, Monolaurin, Protocatechuic acid, Daumone, Cyanidin 3-(2G-glucosylrutinoside), Astragalin, Palmitic acid, β-Sitosterol. But in the contrary, 4-O-Caffeoylquinic acid, Quercetin-3-(6’’-malonyl)-Glucoside and Syringic acid are prominent in the plant extract that the product. Some compounds were noticed to be in minute concentarion or even absent in product in market like Avicularin, Hydroxyphloretin-2’-O-β-glucoside, Luteolin 7-(6’’-malonylneohesperidoside) and Rutin that might be due to loss during processing or even suggesting degradation as Quercetin 3-robinobioside-7-glucoside due to its instability. Methyl galbanate and Chlorogenic acid are completely absent in S2 may could be their sensitivity to the processing conditions. As noticed in S2, Magnesium stearate and Tralonide and Indigotine FD&C Blue 2 additives that appeared in high concentration in the product only, the first one is a synthetic corticosteroid that added to enhance the bioavailability as it is considered as flow agent that inhibits the sticking of ingredients inside the capsule27 while tralonide is used as synthetic corticosteroids used to enhance the anti-inflammatory potentials28. The last one plays a role as colorant used in drug dosage, food and cosmetics29.
Cytotoxicity
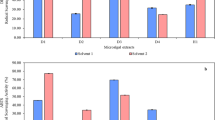
This study screened the cytotoxic activities (IC50 values) of two samples: Saw palmetto crude extract S1 and the product S2 compared to standard Staurosporine against two prostate cancer cell lines (PC3 and DU-145). The results revealed that both samples under investigation offered cytotoxic activity, with some differences in their potency against the mentioned cell lines. The product in market showed significantly lower IC50 values compared to the crude saw palmetto extract, reflecting higher cytotoxic activity. Regarding PC3 cells, IC50 of the product was 7.30 ± 0.32 µg/ml while the extract’s IC50 was 20.04 ± 0.89 µg/ml compared to standard staurosporine (IC50: 4.44 ± 0.2 µg/ml), which in advance indicates that both samples are still displayed promising activity. Similarly, in DU-145 cells, the product (IC50: 36.64 ± 1.97 µg/ml) showed improved cytotoxicity than the crude extract (IC50: 50.74 ± 3.06 µg/ml) and IC50: of the standard was 25.03 ± 1.41 µg/ml. These findings confirmed that the product in market may possess enhanced therapeutic potential compared to the crude extract, with an activity approaching the commonly reported anticancer agent staurosporine. The finding is summarized in Table 2 and figure S3.
In-silico docking and molecular simulation
Molecular docking validation
In this study, we targeted the identified compounds of greatest prevalence in LC-MS data. The x-ray crystal structure of CDK2 (PDB:3TI1), which boasts complete protein coverage and a resolution of 1.99 Å, was employed to investigate the binding of ten saw palmetto identified compounds; Rutin (C11), Quercetin-3-(6’’-malonyl)-Glucoside (C12), Methyl galbanate (C19), β-sitosterol 3-O-glucoside (C34), Luteolin 7-(6’’-malonylneohesperidoside) (C37), Quercetin 3-robinobioside-7-glucoside (C38), Cyanidin 3-(2G-glucosylrutinoside) (C39). 3-Ara-28-Glu Hederagenin (C41), Syringic acid (C46) and 2-Hydroxycinnamic acid (C47), Fig. 1. The choice of the docking site and the precision of the docking calculations were confirmed by re-docking the co-crystalized ligand (sunitinib). The re-docking of the ligand resulted in a slight deviation, with a Root Mean Square (RMS) value of 0.60 Å, demonstrating the reliability of the docking analysis (Figure S4).

Structures of major metabolites in samples under investigation.
Molecular docking studies
The molecular docking analysis demonstrated that most of the selected saw Palmetto extracted natural compounds exhibited good binding within the active site of CDK2, establishing crucial interactions with the ATP-binding pocket residues. The binding poses of the top-scoring ligands were analyzed in detail to assess their binding affinity, stability, and key intermolecular interactions. The extra precision (XP) docking modes were employed to obtain more reliable docking results.
Protein-ligand interactions
A consistent feature among the docked inhibitors was hydrogen bonding to the CDK2 hinge region – specifically, H-bonds with the backbone –NH of LEU83 and the carbonyl of GLU81. These hinge interactions are critical for stabilizing inhibitors in the ATP site. Ligands forming these H-bonds generally had better docking scores. In addition to polar contacts, most docked compounds made hydrophobic interactions with key active-site residues (VAL18, VAL64, ALA31, LYS33, PHE80, etc.), occupying the hydrophobic pocket near the ATP-binding site. The ligands positioned their aromatic and nonpolar moieties to maximize van der Waals contacts with these residues, further stabilizing the complex.
One of the highest-scoring molecules, Compound 12, demonstrated strong hydrogen bonding interactions with GLU 81 and LEU 83, along with additional hydrogen bonds to LYS 89, ASN 132, and ASP 145 (Fig. 2). The involvement of ASN 132 in polar interactions, though less common, suggested that Compound 12 had an extended interaction profile, which contributed to its superior binding affinity. The docking score for Compound 12 was − 13.971 kcal/mol in XP mode, reflecting strong binding potential compared to the controls sunitinib and staurosporine which exhibited binding affinity scores of − 11.437 kcal/mol and − 2.806 kcal/mol, respectively. Both Compound 12 (quercetin-3-(6’’-malonyl)-glucoside) and Compound 37 (luteolin 7-(6’’-malonylneohesperidoside)) formed the crucial hydrogen bonds with the CDK2 hinge residues LEU83 and GLU81 – a common motif for ATP-site inhibitors that helps explain their favorable docking scores. They also shared hydrophobic contacts with key ATP-pocket residues (e.g., VAL18, PHE80, LEU134; see Table 3). However, notable differences were observed in their interaction profiles. Compound 12 engaged in an extended polar network, forming additional hydrogen bonds with LYS89, ASN132, and ASP145. In contrast, Compound 37 did not interact with ASN132 but instead maintained a particularly persistent hydrogen bond with ASP145 and made an extra contact with GLU162 during the MD simulations. Thus, Compound 12’s high affinity appears to derive from its broader hydrogen-bonding network (including the uncommon ASN132 contact), whereas Compound 37 achieves similarly strong binding through a stable bidentate hinge interaction and a deeper hydrophobic insertion toward GLU162 (as visualized in Fig. 2C). By comparing these top ligands side-by-side, it’s clear each employs a distinct but effective strategy to bind CDK2. The overall hydrophobic and hydrophilic interactions profiles as well as the binding affinity scores are detailed in the Table 3. Intermolecular interactions between docked ligands and CDK2 are shown in two dimensions (right) and three dimensions (left) (PDB: 3TI1) of Compounds 12, 37 and Sunitinib are shown in Fig. 2.

Three-dimensional (left) and two-dimensional (right) depictions of the intermolecular interactions between docked ligands and CDK2 (PDB: 3TI1). (A) Sunitinib, (B) 12 and (C) Compound 37.
Induced fit docking analysis
The IFD approach was employed to assess the flexibility of the Cyclin-Dependent Kinase 2 (CDK2) active site and how it adapts to ligand binding using the Maestro-Schrödinger suite. IFD allows both the ligand and the receptor to undergo conformational adjustments, providing a more realistic view of binding modes compared to rigid receptor docking. According to the obtained IFD scores, all compounds show comparable results; however, the saw palmetto-extracted molecules scored better than the controls. The lowest, highest and average IFD scores for the docked compounds are listed in Table 4.
QM/MM analysis
Subsequent to identifying optimal ligand binding orientations via induced fit docking procedures, rigorous hybrid QM/MM computations were pursued for energetic elucidations with enhanced accuracy. In this composite approach, the CDK2 active site was modeled quantum mechanically while embedding the larger protein system through a molecular mechanic’s force field, balancing precision and computational demands by restricting intensive QM calculations to the reaction center. QM calculations enable direct quantification of molecular attributes related to reactivity that dictate biochemical mechanisms, including frontier molecular orbital parameters such as the highest occupied (HOMO) and lowest unoccupied (LUMO) orbitals signifying ionization potential and electron affinity respectively. The spatial and energetic dispositions between these frontier electron shells influence propensity for electrostatic and covalent interactions. Additionally, the gap between HOMO and LUMO eigenstates denotes electron cloud polarization, with smaller separations corresponding to heightened polarizabilities and chemical reactivities. Total QM/MM interaction energies along with frontier molecular orbital (FMO) parameters were calculated from the optimized geometries in Table 5.
The total QM/MM interaction energies corroborate the relative rankings of the screening hits compared to sunitinib observed in molecular docking. The most stable complex based on QM/MM energy was Compound 6 at −405.25 kcal/mol, followed by Compound 4 and sunitinib. Thus our lead depsidone derivatives formed enhanced noncovalent contacts with the CDK2 binding pocket constituents. Additionally, the ligands display comparable HOMO levels around − 0.3 Hartree, associated with electron donating propensity. Slightly variated LUMOs and conserved gap energies on the order of 0.28 kcal/mol indicate analogous charge transfer capacities across this ligand series. Overall, the QM/MM metrics corroborate the docking affinities and support compounds 4 and 6 as viable CDK2 inhibitor candidates for their balanced electronic distribution and energetic stabilization of the bound CDK2 complexes.
Molecular dynamics results
MD simulations were conducted on the CDK2-ligand complexes from the prior docking studies to evaluate the stability and dynamics of the systems. The complexes were built from the induced-fit docking poses and soaked in orthorhombic water boxes with buffering ions. Desmond was utilized to perform 200 ns simulations under constant NPT conditions at 300 K and 1 atm.
Analysis of the MD trajectories illuminated key structural and energetic trends across the simulation timeframe. The complexes maintained stable temperature, pressure, energy, and volume profiles without abnormal deviations. The ligands remained tightly bound in the ATP-site through conserved hinge region hydrogen bonding and hydrophobic contacts. The backbone RMSDs plateaued around 1-1.8 Å after initial equilibration, signifying the global structures attained steady conformational states. Monitoring of secondary structures indicated the α-helices and β-sheets retained proper folding for functional integrity. Furthermore, radius of gyration and solvent accessible surface area plots confirmed the systems adopted compact, folded states.
To detail ligand-protein interactions, the number of hydrogen bonds and close contacts were computed over the trajectory snapshots. Most of the ligands maintained an average of 2–3 backbone hydrogen bonds with LEU 83 and ASP 86, supplemented by the bridging water molecule. The top scoring compound, compound 12, formed two enduring hydrogen bonds with LEU 83 that were maintained for approximately 65% of the 200 ns molecular dynamics (MD) simulation trajectory. Additional hydrogen bonds were observed between compound 12 and the ASP 86 and ASP 145 residues, persisting for 46% and 36% of the simulation, respectively. Besides these anchored polar contacts, compound 12 established consistent hydrophobic interactions with the PHE 80, LEU 83, and LEU 134 pockets lining the CDK2 binding site (Fig. 3).

Analyses of ligand-protein interactions over the molecular dynamics simulation trajectory. (A) Schematic diagram depicting detailed polar and hydrophobic contacts between the atoms of compound 12 and surrounding CDK2 binding site residues. Key stabilizing interactions are labeled. (B) Normalized stacked bar chart quantifying the per-residue interaction analysis of compound 12 with CDK2 over the simulation timeframe. The chart depicts the fraction of simulation frames where residues make hydrophobic contacts (violet), hydrogen bonds (green) and water bridges (blue) with the ligand.
The preservation of these numerous ligand-receptor interactions over the nanosecond simulation confirms the stability of compound 12 within the CDK2 binding pocket. The hydrogen bonds with LEU 83 and interconnected water network likely constitute pivotal binding elements that will guide future hit-to-lead optimization efforts.
Compound 37 formed persistent hydrogen bonds with key CDK2 residues GLU 81, LEU 83, ASP 145, and GLU 162. Specifically, the hydrogen bonds with LEU 83 and ASP 145 were maintained for over 90% of the 200 ns trajectory (Fig. 4).

Analyses of ligand-protein interactions over the molecular dynamics simulation trajectory. (A) Schematic diagram depicting detailed polar and nonpolar contacts between the atoms of compound 37 and surrounding CDK2 binding site residues. Key stabilizing interactions are labeled. (B) Normalized stacked bar chart quantifying the per-residue interaction analysis of compound 37 with CDK2 over the simulation timeframe.
For comparison, the cocrystallized sunitinib inhibitor displayed greater hydrogen bonding stability over the timeline of the simulation. Sunitinib formed two sustained hydrogen bonds with LEU 83, with the bond between the ligand carbonyl oxygen and residue persisting for 98% of the simulation time. The second hydrogen bond between the sunitinib pyrrole nitrogen and LEU 83 was present for 47% of the MD run. Additionally, sunitinib formed a very stable third hydrogen bond with GLU 81, occupying 99% of the 200 ns simulation (Fig. 5).

Analyses of ligand-protein interactions over the molecular dynamics simulation trajectory. (A) Schematic diagram depicting detailed polar and nonpolar contacts between the atoms of compound 37 and surrounding CDK2 binding site residues. Key stabilizing interactions are labeled. (B) Normalized stacked bar chart quantifying the per-residue interaction analysis of compound 37 with CDK2 over the simulation timeframe.
ADMET analysis
The molecular docking study evaluated the CDK2 inhibitory potential of a panel of saw Palmetto naturally-derived compounds relative to the cocrystallized sunitinib inhibitor. While two lead hits were prioritized for further optimization based on strong predicted binding affinities and favorable ligand binding modes, Table 6 highlights key physicochemical limitations underlying the depsidone scaffolds. Specifically, the elevated molecular weights (> 500 Da) and high aqueous solubilities (QPlogPo/w > 3) reduce bioavailability, reflected by 0% human oral absorption predictions which can be enhanced, in future work, by prodrug development, encapsulation, liposomes or conjugation with virus-like particles. Moreover, the compounds exhibit high counts of metabolizing groups, with 8–9 sites prone to clearance mechanisms. Hence, while certain depsidones exhibit promising interactions with CDK2 comparable to sunitinib, the scaffolds suffer from suboptimal drug-like properties, namely solubility and permeability that curtail oral absorption and biological activity.
Conclusion
This study investigates the chemical composition, biological activity, and molecular interactions of Serenoa repens seed extract (S1) and a pharmaceutical formulation (S2) containing Serenoa repens. The findings provide insights into the therapeutic potential of these samples, with emphasis on their cytotoxic effects, chemical profiles, and molecular docking outcomes. Phytochemical analysis identified approximately 50 bioactive compounds in both S1 and S2, representing diverse classes of secondary metabolites. This diversity underpins their biological activities, including anti-androgenic effects. These findings position that the superior performance of S2 indicates the advantage of pharmaceutical processing in enhancing efficacy. The Serenoa repens product (S2) demonstrated greater in vitro cytotoxic activity against prostate cancer cells compared to the crude extract (S1), suggesting that the formulation process may enrich or preserve active ingredients. Furthermore, the findings provide a foundation for the rational design of pharmaceutical formulations incorporating Serenoa repens bioactives for improved therapeutic outcomes. When compared to purified product extract, crude saw palmetto extract showed less cytotoxic effects on prostate cancer cells. This could be due to a variety of factors, including the fact that co-occurring compounds like flavonoids and tannins can sometimes modulate the cytotoxic activity by competing for cellular uptake or acting as antagonists30 or competitors that reduce bioavailability and alter the activity31. Additionally, differences in the environmental conditions and plant preparation techniques, such as drying and extraction techniques, may be the reason32. The pharmaceutical processing of S1 into S2 likely altered the concentrations of specific bioactives, enhancing the therapeutic potential of S2, as corroborated by LC-MS data discussed in the broader context of chemical transformations. The MTT cytotoxicity assay demonstrated significant anti-androgenic activity of both S1 and S2 against prostate cancer cell lines (Pc3 and DU-145). Notably, S2 exhibited enhanced cytotoxic effects compared to S1. This superiority may stem from the selective enrichment or stabilization of active compounds during the pharmaceutical formulation process. These results highlight S2 as a more potent candidate for therapeutic applications targeting androgen-dependent pathways in prostate cancer. In Silico docking and Molecular Dynamics Simulations showed that Compound 12 (Quercetin-3-(6’’-malonyl)-Glucoside) and compound 37 (Luteolin 7-(6’’-malonylneohesperidoside) demonstrated strong inhibitory potential against CDK2, with high Glide XP scores indicating favorable binding affinities, further supported by MD simulations that confirmed stable interactions over time. The interaction analysis revealed significant hydrophobic and hydrophilic contacts, suggesting a stable and specific interaction with the CDK2 active site. Compound 12 exhibited the highest inhibitory potential against CDK2 suggesting strong binding affinity and specificity, corroborated by stable interactions during molecular dynamics simulations, it was recently proved its antioxidant33 and anti-inflammatory potentials34. Compound 37 showed a slightly lower but still significant docking score, indicating substantial inhibitory activity against CDK2. Both compounds demonstrated stable interactions with CDK2 throughout the MD simulations, which reinforces their potential as effective inhibitors. Overall, as potent natural inhibitors of CDK2. The findings provide a basis for further development of Serenoa repens-based pharmaceuticals to enhance therapeutic efficacy and precision in cancer management. Our in silico results suggest that certain identified phytochemicals (e.g., quercetin-3-malonyl-glucoside and luteolin malonylneohesperidoside) bind strongly to CDK2, indicating their potential as CDK2 inhibitors. This is notable since CDK2 has emerged as an important therapeutic target in advanced prostate cancer16especially in tumors that evade CDK4/6 inhibition35. In fact, a combined CDK2/4/6 inhibitor has recently entered clinical trials35underscoring the translational interest in CDK2-targeted therapies. While our findings highlight promising natural lead compounds for CDK2 inhibition, we stress that these conclusions are based on computational models and cell assays. Therefore, in vivo studies are necessary to confirm the therapeutic efficacy and safety of S2 and its key constituents.
Future directions are recommended including pharmaceutical development: The enhanced bioactivity of S2 suggests a need for further refinement of extraction strategies using response surface methodology (RSM) and formulation techniques to optimize the retention and activity of key bioactives. In-vivo validation; While the in-vitro and in silico findings are promising, in-vivo studies are essential to confirm the therapeutic potential and safety of S2. Mechanistic Studies: Investigating the precise molecular mechanisms by which these compounds exert their anti-androgenic and CDK2-inhibitory effects can provide deeper insights into their therapeutic value. Further studies are warranted to explore the mechanisms underlying its cytotoxic effects and to assess its potential for clinical applications in prostate cancer treatment.
Data availability
Data is provided within the manuscript and supplementary information files.
References
Ørsted, D. D. & Bojesen, S. E. The link between benign prostatic hyperplasia and prostate cancer. Nat. Reviews Urol. 10, 49–54 (2013).
Aggarwal, S. et al. An overview of phytotherapeutic approaches for the treatment of benign prostatic hyperplasia. Mini Rev. Med. Chem. 14, 257–270 (2014).
Xiang, J., Li, Z. & Wang, C. Exploring the mechanism of action of Qian lie Xing Fang during the treatment of benign prostatic hyperplasia via network Pharmacology and molecular dynamics simulation analyses. Medicine 102, e35540 (2023).
Matsukawa, Y. et al. Useful parameters to predict the presence of detrusor overactivity in male patients with lower urinary tract symptoms. Neurourol. Urodyn. 39, 1394–1400 (2020).
Kim, E. H., Brockman, J. A. & Andriole, G. L. The use of 5-alpha reductase inhibitors in the treatment of benign prostatic hyperplasia. Asian J. Urol. 5, 28–32 (2018).
Mohammed, H. A. et al. Phytochemical, biological, and computational investigations of ephedra Alata decne. Growing in salinity conditions of Arabian Peninsula. Sci. Rep. 14, 21987 (2024).
Mando, H., Hassan, A., Abo Chameh, G., Moussa, N. & QUALITY ASSESSMENT OF HERBAL MEDICINAL PRODUCTS IN BENIGN PROSTATE HYPERTROPHY. CHEMICAL MARKERS AND FINGERPRINT ANALYSIS. Bull. Pharm. Sci. Assiut Univ. 46, 983–996 (2023).
Leisegang, K. et al. A systematic review of herbal medicine in the clinical treatment of benign prostatic hyperplasia. Phytomedicine Plus. 2, 100153 (2022).
Liang, Y. Z., Xie, P. & Chan, K. Quality control of herbal medicines. J. Chromatogr. B. 812, 53–70 (2004).
Buonocore, D. et al. Serenoa repens extracts: in vitro study of the 5α-reductase activity in a co-culture model for benign prostatic hyperplasia. Archivio Italiano Di Urol. E Andrologia. 90, 199–202 (2018).
Wyatt, G. K., Sikorskii, A., Safikhani, A., McVary, K. T. & Herman, J. Saw palmetto for symptom management during radiation therapy for prostate cancer. J. Pain Symptom Manag. 51, 1046–1054 (2016).
Braeckman, J. The extract of Serenoa repens in the treatment of benign prostatic hyperplasia: a multicenter open study. Curr. Therapeutic Res. 55, 776–785 (1994).
Marti, G. et al. Comparison of the phytochemical composition of Serenoa repens extracts by a multiplexed metabolomic approach. Molecules 24, 2208 (2019).
Rashid, K., Raj, V., Kumar, P. & Nishad, K. Hair care promising herbs: A review. Pharm. Res. 10, 677–688 (2020).
Anastassakis, K. in Androgenetic Alopecia From A to Z: Vol. 2 Drugs, Herbs, Nutrition and Supplements 429–439Springer, (2022).
Yin, X. et al. Identification of CDK2 as a novel target in treatment of prostate cancer. Future Oncol. 14, 709–718 (2018).
Che, Y., Hou, S., Kang, Z. & Lin, Q. Serenoa repens induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of STAT 3 signaling. Oncol. Rep. 22, 377–383 (2009).
Ismail, A. et al. Chemical composition and therapeutic potential of three Cycas species in brain damage and pancreatitis provoked by γ-radiation exposure in rats. J. Radiation Res. Appl. Sci. 13, 38–52 (2020).
Aati, H. Y. et al. Garcinia cambogia phenolics as potent anti-COVID-19 agents: phytochemical profiling, biological activities, and molecular Docking. Plants 11, 2521 (2022).
Riss, T. L. et al. Cell viability assays. Assay guidance manual [Internet] (2016).
Edmondson, J. M., Armstrong, L. S. & Martinez, A. O. A rapid and simple MTT-based spectrophotometric assay for determining drug sensitivity in monolayer cultures. J. Tissue Cult. Methods. 11, 15–17 (1988).
Bhullar, K. S. et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer. 17, 1–20 (2018).
Martin, M. et al. A novel approach to the discovery of small-molecule ligands of CDK2. Chembiochem 13, 2128–2136 (2012).
Tripathi, S. K., Muttineni, R. & Singh, S. K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 334, 87–100 (2013).
Kim, S. S., Alves, M. J., Gygli, P., Otero, J. & Lindert, S. Identification of novel Cyclin A2 binding site and nanomolar inhibitors of Cyclin A2-CDK2 complex. Curr. Comput.-Aided Drug Design. 17, 57–68 (2021).
Bochevarov, A. D. et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 113, 2110–2142 (2013).
Veronica, N., Loh, Y. Y., Loh, L. X. Y., Heng, P. W. S. & Liew, C. V. Impact of magnesium stearate physical and chemical variabilities on pharmaceutical powder flow and tablet physical properties. J. Pharm. Invest. 54, 209–227 (2024).
Publishing, W. A. Pharmaceutical Manufacturing EncyclopediaVol. 1 (Elsevier, 2007).
Food, U. & Administration, D.
Kota, S. K. & Balasubramanian, S. Cancer therapy via modulation of micro RNA levels: a promising future. Drug Discovery Today. 15, 733–740 (2010).
Chang-Xiao, L. & Pei-Gen, X. Recent advances on ginseng research in China. J. Ethnopharmacol. 36, 27–38 (1992).
Mohammed, H. A. et al. Variability in the volatile constituents and biological activities of Achillea millefolium L. essential oils obtained from different plant parts and by different solvents. Arab. J. Chem. 16, 105103 (2023).
Panat, N. A. et al. Antioxidant profiling of C3 Quercetin glycosides: quercitrin, Quercetin 3-β-D-glucoside and Quercetin 3-O-(6-O-malonyl)-β-Dglucoside in cell free environment. Free Radicals Antioxid. 5, 90–100 (2015).
Terao, J. Potential role of Quercetin glycosides as anti-atherosclerotic food-derived factors for human health. Antioxidants 12, 258 (2023).
Siskin, M., Economides, M. P. & Wise, D. R. Cyclin-Dependent kinase Inhibition in prostate cancer: past, present, and future. Cancers 17, 774 (2025).
Acknowledgements
The work team thank the Deanship of Graduate Studies and Scientific Research at Jouf University.
Funding
This work was funded by the Deanship of Graduate Studies and Scientific Research at Jouf University under grant No. (DGSSR-2023-01-02085).
Author information
Authors and Affiliations
Contributions
Conceptualization, E.M and A.I; methodology, E.M, A.I, A.K, M.A., S.S, H.A; software, A.I., M.A.; validation E.M, A.I., N.A, T.S; formal analysis, A.I., E.M., S.H.; investigation, A.I., E.M., A.K; writing—original draft preparation, E.M, A.I, A.K, M.A.; writing—review and editing, E.M, A.I, A.K, M.A.; All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mostafa, E.M., Aldekhail, N.M., Alnusaire, T.S. et al. Comparative study of the chemical profile, cytotoxic activity, and molecular docking of Serenoa repens extract and a pharmaceutical product. Sci Rep 15, 34338 (2025). https://doi.org/10.1038/s41598-025-16758-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-16758-z
