
Abstract
Hearing loss is genetically heterogeneous, with over 121 implicated genes. Minor allele frequency (MAF) data from population databases greatly aid variant interpretation; however, these databases are predominantly based on individuals of European ancestry and lack sufficient East Asian representation, limiting accurate interpretation in under-represented populations. We analyzed rare variants associated with non-syndromic hearing loss classified as pathogenic, likely pathogenic, or of uncertain significance in the Deafness Variation Database (DVD). Population allele frequencies from 9,579 Koreans, 54,000 Japanese, and 651 patients with sensorineural hearing loss were evaluated. Of the 6,381 pathogenic or likely pathogenic variants cataloged in the DVD, 216 (3.38%) were detected in Korean population. Among these, 31 variants exhibited high allele frequencies that exceeded thresholds typically applied to identify benign variants in clinical interpretation guidelines. Of these, 6 remained disease-causing, including 4 East Asian founder alleles and one MYO7A variant common in Koreans. Our pipeline identified 24 variants for reclassification as benign or likely benign, and one P2RX2 variant of uncertain significance. Of 1,299,211 VUS, 3,736 were reclassified as benign. A substantial number of variants previously classified as pathogenic were reclassified as benign using MAF data from under-represented populations, highlighting the need for large-scale sequencing in diverse ancestries.
Similar content being viewed by others


Introduction
Hearing loss is one of the most prevalent sensory diseases with high genetic heterogeneity. Congenital hearing loss occurs in approximately 1–2 cases per 1,000 newborns1. The prevalence increases with age, affecting over half of individuals aged 85 and older with mild to profound hearing loss1. Hearing loss can occur independently as a non-syndromic condition, accounting for ~ 70% of cases, or alongside other clinical features as part of a syndrome in the remaining 30%2,3. Approximately half of non-syndromic hearing loss (NSHL) have a genetic basis, with > 121 genes linked to monogenic forms of hearing loss. Around 80% of prelingual hearing loss is caused by variants in the genome sequence, most often autosomal recessive and non-syndromic4. Among the various types of disease-causing variants linked to NSHL, missense variants are the most common, followed by indel/frameshift, nonsense variants, and splice site variants5.
Owing to genetic heterogeneity, the accurate interpretation of variants in the molecular diagnosis of hearing loss can be particularly challenging. The Deafness Variation Database (DVD; https://deafnessvariationdatabase.org/) is the largest up-to-date repository for the clinical interpretation of variants associated with hearing loss5. It interprets previously reported and newly identified variants from population databases. The internal pipeline used by DVD, called Kafeen, selects variants based on expert curation compared to other clinical databases (such as HGMD and ClinVar), in silico prediction scores, and the minor allele frequency (MAF) in various population databases.
MAF is considered a key criterion for selecting pathogenic variants and filtering out those that are too common to cause diseases. According to American College of Medical Genetics and Genomics (ACMG) guidelines, the Benign, Stand-alone (BA1) criterion is used to classify variants with high MAF as benign6. Kafeen uses the highest population-specific MAF from major population databases, including 1000 Genomes, Exome Sequencing Project (ESP), The Exome Aggregation Consortium (ExAC), and The Genome Aggregation Database (gnomAD), to determine variant frequencies7,8. To date, 10,987 variants have been classified as pathogenic or likely pathogenic in the DVD, and associated with either syndromic or non-syndromic hearing loss.
However, population-specific classifications of common or rare variants are not readily generalizable, because the data in major population databases are largely from individuals with European ancestry. For example, in gnomAD v4 (released in November 2023), 77.07% of the 807,162 individuals are of European descent; East Asians comprise only 2.78%. The under-representation of ethnically diverse populations in genomic research hinders our understanding of the genetic architecture of human diseases and contributes to health disparities. Specifically, the lack of diversity in population databases can lead to inaccurate MAF estimates for under-represented populations, potentially misclassifying benign variants as disease-causing, resulting in inaccurate molecular diagnoses. Therefore, using a major population database may limit the accurate assessment of variants identified in individuals from under-represented populations.
Koreans, as part of the East Asian population, are one such under-represented group. In gnomAD v2.1.1, only 1,916 Korean individuals were included, and while gnomAD v4 increased the East Asian representation by 2.3 times, the sample size may still be insufficient for accurate variant interpretation. In this study, we aimed to reevaluate the classification of reportedly pathogenic variants by incorporating allele frequencies from a Korean population. We deposited 6,381 pathogenic (P) or likely pathogenic (LP) variants and 1,299,211 variants of uncertain significance (VUS) from 121 genes associated with NSHL in the DVD. By comparing these variants with the allele frequencies of 9,579 Korean and 54,000 Japanese individuals, we identified variants with elevated frequencies in the Korean population as targets for reinterpretation.
Results
Variant interpretation and penetrance estimation
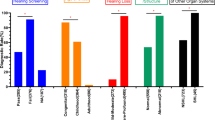
In the DVD, we extracted 6,381 P or LP variants of hearing loss genes. Of these, 216 (3.38%) variants in 121 NSHL genes were identified in a cohort of 9,579 Korean individuals (Fig. 1, Supplementary Table S1). We compared variant allele frequencies with those from two Korean control cohorts: KOVA2, a publicly available genome dataset comprising healthy individuals, and CIRN, a clinical sequencing dataset derived from presumed unaffected individuals (see Methods for details). Among these, 31 variants were identified as high-frequency variants for MAFs calculated from either the KOVA2 cohort (n = 5,305 individuals) or CIRN cohort (n = 4,274 individuals). Through literature searches, we identified 4 of the 31 variants (GJB2 [NM_004004.6]: c.109G > A;p.(Val37Ile)9,10,11, c.109G > A;p.(Leu79Cysfs*3)12,13; SLC26A4 [NM_000441.2]: c.2168A > G;p.(His723Arg)12,14,15; KCNQ4 [NM_004700.4]: c. 140 T > C;p.(Leu47Pro)16,17) as pathogenic founder alleles in East Asian populations (Supplementary Table S2). Evidence for the pathogenicity of these variants has been well established through segregation analyses, mouse models, and cell-based assays.

Flowchart of the reclassification method and results. KOVA2, Korean Variant Archive 2; CIRN, Clinical Interpretation Research Network; YUHL, Yonsei University Hearing Loss cohort; ToMMo, JPN Tohoku Medical Megabank Organization. P/LP: pathogenic or likely pathogenic variants.
We reevaluated the remaining 27 variants using allele frequency (AF) data from ToMMo JPN (n = ~ 54,000)18,19,20, a population dataset representing the Japanese population, which is genetically similar to the Korean population and under-represented in gnomAD (n = 76) (Supplementary Table S3)8. Following the ACMG guidelines adapted for hearing loss, we applied the Benign, Stand-alone criterion (BA1; MAF ≥ 0.001 for dominant genes and MAF ≥ 0.005 for recessive genes) and the Benign, Strong 1 criterion (BS1; MAF ≥ 0.0002 for dominant genes) criteria to filter variants with frequencies too high to be pathogenic. To apply a more conservative filter, we only assigned BA1 or BS1 criteria if variants satisfied the MAF cutoffs in at least two of the three population databases (KOVA2, CIRN, and ToMMo JPN), as described in Supplementary Table S4.
Among the 27 high-frequency variants, we assigned the BA1 criterion to 14 variants and the BS1 criterion to 12 variants (Supplementary Table S3). Consequently, 26 variants were found too common in the under-represented population to be considered plausible causes of hearing loss. However, we cannot rule out the possibility that these high-frequency variants represent newly introduced pathogenic founder alleles. Therefore, we referred to AF in a case cohort consisting of exome and genome sequence data from 651 patients (Fig. 1)21,22,23,24. We identified 16 of the 26 variants in the case cohort, mostly at very low frequencies, and strengthened the reclassification confidence for 10 variants (Supplementary Figure S1). We further tested the pathogenicity of these high-frequency variants through segregation analysis and penetrance estimation, as detailed in the Methods section.
Segregation analysis and penetrance estimation
With the exception of P2RX2 (NM_170683.4): c.817G > T; p.(Asp273Tyr) and MYO7A (NM_000260.4): c.2023C > T; p.(Arg675Cys), most high-frequency variants did not exhibit clear segregation consistent with disease. Some showed partial co-segregation—for example, TECTA p.(Thr165Ile) and TJP2 p.(Thr1188Ala)—but the evidence was insufficient to meet the PP1 criterion under ACMG guidelines (Supplementary Figure S2). In certain cases, such as MYH9 p.(Arg802Trp) (YUHL364), segregation analysis could not be conducted due to the unavailability of parental samples. Penetrance estimates for 13 autosomal dominant variants indicated a very low risk of hearing loss, with the calculated estimates and their 95% confidence intervals (CIs), falling below baseline risk. As a result, these variants were reclassified as benign or likely benign (Supplementary Table S3 and Fig. 2).

Lifetime risk of P/LP variants frequently detected in Korean patients. Mean penetrance values are shown with 95% confidence intervals for each variant based on data from three East Asian population databases (two Korean and one Japanese). The vertical dashed line represents the baseline lifetime risk of hearing loss in the general population (3.3%). Variants are ordered by their estimated penetrance. Error bars reflect the variability of penetrance estimates across population datasets.
The mean penetrance estimate for the MYO7A p.(Arg675Cys) variant was 0.19 [95% CI; 0.04–0.77], higher than the baseline risk for late-onset hearing loss (0.033) [Supplementary Table S3]. In addition, odds ratio (OR) analyses using CIRN and ToMMo control cohorts supported the association of this variant with late-onset hearing loss, yielding a statistically significant OR (lower 95% CI > 1 and P < 0.05; Supplementary Table S6). While the AF of this variant was higher than the BS1 cutoff in two Korean control groups (KOVA2 and CIRN), it has been causally linked to late-onset hearing loss25 and is extremely rare in all subpopulations of gnomAD, including East Asian populations (Supplementary Table S5). According to family based penetrance calculations, 88% of individuals carrying the MYO7A variant develop hearing loss by the age of 50 (Supplementary Figure S3). Given its segregation in families with late-onset hearing loss and its relatively common presence in the Korean population, we considered the MYO7A p.(Arg675Cys) variant as a potentially deleterious founder allele and reclassify it as likely pathogenic. The estimated penetrance for the P2RX2 p.(Asp273Tyr) variant was 0.05 [95% CI; 0.016–0.19]. The lower bound (95%) was lower than the baseline risk for late-onset hearing loss. Although the AF of the P2RX2 variant exceeded the BA1 criterion of 0.001 in all three population databases (KOVA2, CIRN, and ToMMo), this variant showed familial segregation with late-onset hearing loss (Supplementary Figure S4) and was functionally characterized as a likely cause of hearing loss26,27,28. Based on this evidence, we reinterpreted the variant as a variant of uncertain significance (VUS) that requires further functional characterization to establish a robust phenotype–genotype correlation.
Reinterpretation and impact
We applied a similar reinterpretation scheme to VUSs. DVD houses 1,299,211 VUS, of which 38,107 (2.93%) were identified in the KOVA2 and ToMMo54K population databases. We confirmed that 3,736 variants in 121 NSHL genes were common in both the KOVA2 and ToMMo databases, exceeding the MAF cutoff for the BA1 criteria (≥ 0.5% in AR or AR/AD genes or ≥ 0.1% in AD genes), and therefore reclassified them as benign variants. The reinterpretation of variants led to significant shifts in the classification of 11.57% (25/216) of P/LP variants and 9.80% (3,736/38,107) of VUS variants (Fig. 3), except for reclassification of MYO7A p.(Arg675Cys) variant which was reclassified from P to LP. In the case of the reclassified P/LP variants, one variant changed from P to VUS, one from P to LP, four changed from P to LB, six changed from P to B, seven initially classified as LP changed to LB, and seven to B (Fig. 3). Therefore, we reclassified 9.82% (3,762/38,323) of the P/LP/VUS variants by examining their MAF in a large-scale under-represented population database.

Reclassification of hearing loss-associated variants based on under-represented population databases. Sankey plot showing reclassification of HL-associated variants using Korean allele frequency data. Variants originally classified as P, LP, or VUS (left) were reassessed and reclassified into ACMG categories (right). Stream widths reflect variant counts (√-scaled). Many VUS were reclassified as benign, underscoring the value of population-specific data.
As shown in Table 1, 27 high-frequency P/LP variants were localized to 14 genes: MYH14 (6 variants), TECTA (5 variants), TJP2 (3 variants), MYH9 and WFS1 (2 variants each), and P2RX2, KCNQ4, TRIOBP, DIAPH1, GSDME, GRHL2, MYO6, MYO7A, and EYA4 (1 variant each). Among these, 26 variants were reclassified, and all exonic variants were associated with NSHL. Of the 26 reclassified variants, 25 were missense, and one variant of GSDME was a nonsense variant.
To evaluate the concordance between our reclassification and publicly available interpretations, we compared out updated classifications with ClinVar annotations (Supplementary Table S3). Of the 27 high-frequency variants, 20 were present in ClinVar. Most of these were listed as VUS, often submitted by a single source, and eight displayed conflicting interpretations (e.g., VUS vs. LB or P vs. VUS). These findings underscore the value of incorporating population-specific AF data to help resolve uncertain or discrepant variant classifications.
Diagnostic rate in population controls
After reinterpreting the pathogenic variants, we further characterized the potential diagnostic rate for NSHL in the Korean population using the analysis pipeline described in Supplementary Figure S5. To avoid inflation of the total diagnostic rate, we calculated the rates with and without potential founder alleles in the dominant genes, such as the p.(Leu47Pro) variant in KCNQ4 and p.(Arg675Cys) variant in MYO7A (Table 2). For P/LP variants in recessive genes, we included individuals with homozygous P/LP variants or P/LP variant pairs in the double-heterozygous (potentially compound) state. Based on the trio data from the CIRN, we filtered the variant pairs in the cis configuration when possible.
As a result, 26 of 5,305 individuals in KOVA2 (0.49%) and 26 of 4,274 individuals in CIRN (0.60%) carried rare disease-causing variants in dominant inheritance, whereas 17 of 5,305 individuals (0.32%) in KOVA2 and 14 of 4,274 individuals (0.32%) in CIRN carried recessive variants in potential pathogenic combinations (Table 2). Excluding p.(Leu47Pro) variant of KCNQ4 and p.(Arg675Cys) variant of MYO7A, missense variants of MYO6 were the most common causes of hearing loss in the dominant genes, whereas genetic variants of MYO15A were predominant in the recessive genes, followed by GJB2 and SLC26A4 (Supplementary Table S7). On average, 0.87% of individuals in the Korean population exhibit a genetic predisposition to develop NSHL, with 0.81% in KOVA2 and 0.93% in CIRN. This condition manifests across a wide range of disease onset stages.
Discussion
The accurate interpretation of genetic variants remains a significant challenge in clinical genetics, particularly for genetically heterogeneous disorders such as NSHL. Whereas population AF is a key criterion in variant interpretation guidelines, existing population databases are skewed toward European ancestry, which limits the applicability of AF thresholds to other populations. In this study, we addressed this disparity by integrating large-scale AF data from under-represented East Asian populations, specifically Korean and Japanese populations, to assess the pathogenicity of hearing loss-associated variants. By comparing AFs from 9,579 Korean controls (KOVA2 and CIRN) and 54,000 Japanese individuals (ToMMo 54KJPN) with variant classifications from the DVD, we identified 31 variants that exceeded AF thresholds used in the ACMG guidelines. Among these, 6 variants remained classified as disease-causing owing to compelling evidence from segregation, mouse models, or high penetrance in the patient cohort. Notably, we identified the p.(Arg675Cys) variant of MYO7A as a likely pathogenic variant and as a novel founder variant in the Korean population.
Of particular clinical significance, 24 of the 31 high-frequency variants were reclassified as benign or likely benign, and 1 variant, P2RX2 p.(Asp273Tyr), was reinterpreted as a VUS owing to conflicting evidence from AF and functional studies. Notably, previous studies have reported associations between P2RX2 variants and both autosomal dominant progressive hearing loss and increased susceptibility to noise-induced hearing loss. These findings may help explain the observed segregation and functional relevance of the p.(Asp273Tyr) variant, despite its relatively high AF29,30. These reclassifications are directly relevant to the clinical interpretation of genetic test results and counseling of patients and families, preventing misdiagnoses based on outdated or population-inappropriate variant classifications. Furthermore, by evaluating pathogenic variant combinations in dominant and recessive genes, we estimated that approximately 0.87% of individuals in the general Korean population may harbor pathogenic variants linked to NSHL. This study provides a valuable reference for assessing background carrier frequencies in population screening and incidental findings in genome sequencing. Our findings reinforce the importance of using ethnically matched population data to interpret variants. The overrepresentation of certain variants in East Asian populations led to their misclassification as pathogenic in earlier studies that relied solely on European datasets. This highlights the critical need for increased diversity in reference databases and supports a growing movement toward population-specific interpretation frameworks.
This study had several limitations. First, the lack of auditory phenotyping in the control datasets introduces uncertainty in the penetrance estimates. Second, although segregation and population data were available, functional validation was not conducted for all the reclassified variants. In addition, while the Korean and Japanese populations share considerable genetic similarity, subtle differences in AF and genetic architecture between subgroups may still affect the interpretation. Despite these limitations, our study provides a strong example of how integrating under-represented population data can refine the accuracy of variant interpretations. As genomic sequencing becomes more prevalent globally, efforts to expand the diversity of reference datasets must be prioritized. Future research should also include longitudinal follow-up of individuals with high-frequency variants and functional studies to further delineate genotype–phenotype correlations, particularly for VUS, such as P2RX2 p.(Asp273Tyr). In conclusion, our study demonstrated that leveraging data from under-represented East Asian populations can substantially improve the classification of hearing loss-associated variants. This approach enhances diagnostic precision and contributes to equitable genomic medicine by reducing health disparities caused by under-representation in genomic research.
Methods
Research subjects
We used two distinct population datasets as controls to extract pathogenic variants and their MAFs. The Korean Variant Archive 2 (KOVA2) and Clinical Interpretation Research Network (CIRN) were the two datasets. KOVA2 is a ready-to-use dataset composed of exome and genome sequencing data from 5,305 Korean individuals. It provides a single VCF file containing variants, including MAFs, homozygous variant counts, and allele numbers, which are stored in the ‘KOVA_AF,’ ‘KOVA_Hom_Count,’ and ‘KOVA_AN’ fields in the ‘INFO’ section. KOVA2 VCF with genotype information was downloaded from Google Bucket, which was linked to a previous publication31.
Genome sequencing data (gVCF format) for 4,534 controls in the CIRN cohort were obtained from the National Project of Bio Big Data under a controlled access agreement. The controls were from three separate cohorts: 2,500 individuals from the Korean Genome and Epidemiology Study (KoGES), 1,885 from the Rare Disease (RD) cohort, and 149 from the autism spectrum disorder (ASD) cohort. Participants in the KoGES cohort were considered healthy after completing questionnaires assessing 199 health criteria; however, auditory function was not among the evaluation parameters. In the RD cohort, only five individuals had a history of hearing or ear disorders. The ASD cohort consisted of patients with autism spectrum disorders or their family members who consented to participate.
To avoid biasing the MAFs, we created a VCF from 4,534 selected controls and included only 4,274 unrelated individuals, as indicated by the ped file. To obtain MAFs from other under-represented East Asian populations, we used jMorp (https://jmorp.megabank.tohoku.ac.jp/), an online database managed by Tohoku Medical Megabank Organization (ToMMo)32. We referenced AF from the ‘ToMMo 54KJPN’ dataset, which includes data from over 54,000 Japanese controls.
The case cohort included sequencing data from 651 probands with sensorineural hearing loss, excluding individuals with congenital cytomegalovirus infections or other medical conditions primarily affecting hearing function. The majority of cases were NSHL. Of the total, 217 individuals (33.3%) had prelingual onset, while 434 (66.7%) had postlingual onset. Audiometric evaluation revealed that 57 (8.81%) had mild hearing loss, 320 (49.18%) moderate, 99 (15.16%) severe, and 175 (26.84%) profound hearing loss. Based on pedigree data and clinical assessment, 230 probands (35.33%) were presumed to have AD inheritance and 421 (64.67%) AR inheritance. Written informed consent was obtained from all participants. The study was approved by the Institutional Review Board of Severance Hospital, Yonsei University Health System (IRB #4–2015-0659), and all methods were performed in accordance with the relevant guidelines and regulations, including the Declaration of Helsinki.
Variant calling
Genomic DNA was extracted from the blood or saliva samples of 651 patients. Sequencing libraries were prepared using a TruSeq DNA Nano Library Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. We designed a sequence analysis pipeline based on GATK Best Practices for germline variant calling. FASTQ files generated on the Novaseq platform (Illumina, San Diego, USA) were aligned to the hg38 reference genome using the BWA–MEM aligner (v0.7.17)33. After marking duplicates and sorting using MarkDuplicates, the mapping quality was recalibrated using BQSR in GATK (v4.1.9.0)34. Single nucleotide variants (SNVs) and small insertions and deletions (indels) were called for each sample using GATK HaplotypeCaller with the “-ERCGVCF” option35.
For the CIRN cohort, genome sequencing data from 4,534 individuals were deposited as gVCF aligned to hg38. To jointly genotype the samples, we created genomic databases (GenomicsDB) for each chromosome using the GenomicsDBImport in GATK. The SNVs and indels were recalibrated using the VQSR model to select 99.9% and 99.0% of the true sites, respectively, from the training set. The GenomicsDB for CIRN was generated by specifying the ‘interval’ options within the exonic regions of 121 known deafness-associated genes. Variants with GQ < 20 or DP < 10 were filtered using variant filtration. The resulting VCF were annotated using ANNOVAR (https://annovar.openbioinformatics.org/en/latest/)36 and P and LP variants in 121 hearing loss genes were extracted using custom Python scripts.
KOVA2 provides a VCF with pre-calculated AF for each variant. We extracted P, LP, and VUS variants directly from the VCF and referenced the MAFs annotated in the file.
Variant classification deposit
We obtained variants associated with hearing loss from DVD v9 (https://deafnessvariationdatabase.org/)5. Last updated in 2019, the database evaluates variants in 223 deafness-associated genes, classifying them as pathogenic or likely pathogenic (P/LP; 20,895 variants), benign or likely benign (B/LB; 246,577 variants), or of uncertain significance (VUS; 2,226,255 variants). To annotate the VCF and search for variants listed in the DVD, we created a BED file consisting of the following columns: ‘Start,’ ‘End,’ ‘Ref,’ ‘Alt,’ ‘DVD_PATHOGENICITY,’ and ‘DVD_FINALDISEASE.’ This BED file was then imported into ANNOVAR as one of the annotation databases, using distributed Perl scripts such as ‘index-annovar.pl’ and ‘convert2annovar.pl’36.
Variant interpretation
Variant interpretation followed the ACMG guidelines adapted for hearing loss6,37,38. The largest allele numbers for KOVA2 and CIRN were 10,610 and 8,548, respectively, indicating that these two population databases provided reliable sample sizes for calculating population AFs. To define “high-frequency” P/LP variants, we selected those with an allele frequency exceeding 0.5% in AR genes or 0.05% in AD genes in at least one of the Korean control cohorts (KOVA2 or CIRN). For genes associated with both inheritance modes, we applied the threshold corresponding to the reported mode of inheritance for each specific variant. Final reclassifications were made based on the ACMG guidelines with an adjusted criterion for hearing loss. The BA1 cutoffs for variants in dominant and recessive genes were equivalent to AFs suggested in the guideline: MAF of ≥ 0.005 (0.5%) for autosomal recessive and ≥ 0.001 (0.1%) for autosomal dominant genes. We applied the BS1 criterion to the autosomal dominant variants with MAF exceeding 0.02%. To ensure conservative interpretation, the BS1 criterion was not applied to variants in AR genes. We assigned the BA1 or BS1 criteria only if the variant exceeded the thresholds in at least two of the three population databases (KOVA2, CIRN, and ToMMoJPN54K).
In addition to the criterion for population AF, the following ACMG criteria were assigned using VIP–HL (http://hearing.genetics.bgi.com/): BP4 (variants with Rare Exome Variant Ensemble Learner (REVEL) score ≤ 0.15 or predicted to have no impact on splicing in MaxEntScan), PP3 (REVEL score ≥ 0.7 or predicted impact on splicing using MaxEntScan), PP1 (segregation in affected relatives for recessive and two affected relatives for dominant), PP1_Strong (segregation in three affected relatives for recessive and five affected relatives for dominant), PS3_Supporting (functional studies with limited validation show a deleterious effect), PM2_Supporting (filtering POPMAX AF within the threshold 0 ≤ AF < 0.00002 for autosomal dominant variants), PS4_Supporting (autosomal dominant: ≥ 2 probands with the variant and variant meets PM2)39.
Penetrance estimation
For high-frequency variants observed in both the patient cohort and at least two of the three population databases, we estimated penetrance and its 95% confidence interval (CI) using an internal R script based on methods described in previous studies40,41. The calculated mean penetrance estimates across the three datasets were plotted using GraphPad Prism v8.0.2. Penetrance was calculated using case prevalence, observed allele count (AC) in cases, number of alleles sequenced in cases, observed AC in the population, and number of population alleles sequenced (AN)42. The prevalence of late-onset hearing loss was set as 0.033, corresponding to the reported prevalence of hearing loss with onset after age 40, as described in a previous publication3.
Data availability
All relevant data are included in the main manuscript and supplementary materials. The variant-level reclassification results and the R script for penetrance estimation across multiple populations is publicly available at https://github.com/syjoo2304/reclassification.git. Genome sequencing data from the CIRN cohort (4,534 individuals) used in this study were obtained under controlled access as part of the National Project of Bio Big Data, Republic of Korea. These data are not publicly available due to legal and ethical restrictions, as access is restricted to secure analysis environments rather than local downloads. Researchers interested in accessing these data may apply through the National Project of Bio Big Data (https://www.biobigdata.kr/bbd). No new sequencing data were generated for the CIRN or KOVA2 cohorts in this study. The KOVA2 dataset was accessed via the public Google Cloud bucket described in the original publication (Reference #29). All sequencing data for the case cohort (651 patients) were generated in this study but are not publicly available due to privacy and consent limitations. Requests for access to the case cohort data should be directed to the corresponding author and will require IRB approval.
References
Jang, S. H., Yoon, K. & Gee, H. Y. Common genetic etiologies of sensorineural hearing loss in Koreans. Genom. Inform 22, 27. https://doi.org/10.1186/s44342-024-00030-3 (2024).
Vos, B., Noll, D., Pigeon, M., Bagatto, M. & Fitzpatrick, E. M. Risk factors for hearing loss in children: A systematic literature review and meta-analysis protocol. Syst. Rev. 8, 172. https://doi.org/10.1186/s13643-019-1073-x (2019).
Haile, L. M. et al. Hearing loss prevalence and years lived with disability, 1990–2019: findings from the Global Burden of Disease Study 2019. The Lancet. 397, 996–1009 (2021).
Schrijver, I. Hereditary non-syndromic sensorineural hearing loss: transforming silence to sound. J. Mol. Diagn. 6, 275–284. https://doi.org/10.1016/S1525-1578(10)60522-3 (2004).
Azaiez, H. et al. Genomic landscape and mutational signatures of deafness-associated genes. Am. J. Hum. Genet. 103, 484–497. https://doi.org/10.1016/j.ajhg.2018.08.006 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74. https://doi.org/10.1038/nature15393 (2015).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. https://doi.org/10.1038/s41586-020-2308-7 (2020).
Kelley, P. M. et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am. J. Hum. Genet. 62, 792–799. https://doi.org/10.1086/301807 (1998).
Pang, X. H. et al. Characterization of spectrum, rate and genotype-phenotype correlation of dominant mutations in chinese hans. PLoS ONE 9, e100483. https://doi.org/10.1371/journal.pone.0100483 (2014).
Tabor, H. K. et al. Pathogenic variants for mendelian and complex traits in exomes of 6,517 European and African Americans: Implications for the return of incidental results. Am. J. Hum. Genet. 95, 183–193. https://doi.org/10.1016/j.ajhg.2014.07.006 (2014).
Lazarin, G. A. et al. An empirical estimate of carrier frequencies for 400+causal Mendelian variants: Results from an ethnically diverse clinical sample of 23,453 individuals. Genet. Med. 15, 178–186. https://doi.org/10.1038/gim.2012.114 (2013).
Yao, J., Lu, Y. J., Wei, Q. J., Cao, X. & Xing, G. Q. A systematic review and meta-analysis of 235delC mutation of gene. J. Transl. Med. 10(1), 136. https://doi.org/10.1186/1479-5876-10-136 (2012).
Asakura, Y. et al. A patient with pendred syndrome whose goiter progressed with normal serum thyrotropin and iodine organification. Am. J. Med. Genet. A 152(7), 1793–1797. https://doi.org/10.1002/ajmg.a.33456 (2010).
Dai, P. et al. Molecular etiology of hearing impairment in inner mongolia: Mutations in gene and relevant phenotype analysis. J. Transl. Med. 6, 74. https://doi.org/10.1186/1479-5876-6-74 (2008).
Jung, J. et al. Rare KCNQ4 variants found in public databases underlie impaired channel activity that may contribute to hearing impairment. Exp. Mol. Med. 51, 1–12 (2019).
Shin, D. H. et al. A recurrent mutation in KCNQ4 in Korean families with nonsyndromic hearing loss and rescue of the channel activity by KCNQ activators. Hum. Mutat. 40, 335–346. https://doi.org/10.1002/humu.23698 (2019).
Fuse, N. et al. Establishment of integrated biobank for precision medicine and personalized healthcare: The Tohoku Medical Megabank Project. JMA journal 2, 113–122 (2019).
Yasuda, J. et al. Genome analyses for the Tohoku Medical Megabank Project towards establishment of personalized healthcare. J Biochem 165, 139–158. https://doi.org/10.1093/jb/mvy096 (2019).
Nagami, F. et al. Public relations and communication strategies in construction of large-scale cohorts and biobank: Practice in the tohoku medical megabank project. Tohoku J Exp Med 250, 253–262. https://doi.org/10.1620/tjem.250.253 (2020).
Kim, J. A. et al. Systematic genetic assessment of hearing loss using whole-genome sequencing identifies pathogenic variants. Exp Mol Med https://doi.org/10.1038/s12276-025-01428-x (2025).
Joo, S. Y. et al. Bi-allelic variants of SEMA3F are associated with non-syndromic hearing loss. Mol. Cells 48(3), 100190. https://doi.org/10.1016/j.mocell.2025.100190 (2025).
Jung, J. et al. MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss. Exp Mol Med 56, 2423–2435. https://doi.org/10.1038/s12276-024-01338-4 (2024).
Koh, Y. I. et al. OSBPL2 mutations impair autophagy and lead to hearing loss, potentially remedied by rapamycin. Autophagy 18, 2593–2614. https://doi.org/10.1080/15548627.2022.2040891 (2022).
Miyagawa, M., Naito, T., Nishio, S., Kamatani, N. & Usami, S. Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PLoS ONE 8, e71381. https://doi.org/10.1371/journal.pone.0071381 (2013).
George, B., Swartz, K. J. & Li, M. F. Hearing loss mutations alter the functional properties of human P2X2 receptor channels through distinct mechanisms. P Natl Acad Sci USA 116, 22862–22871. https://doi.org/10.1073/pnas.1912156116 (2019).
Housley, G. D. et al. Cochlear homeostasis: A molecular physiological perspective on maintenance of sound transduction and auditory neurotransmission with noise and ageing. Curr Opin Physiol 18, 106–115. https://doi.org/10.1016/j.cophys.2020.09.012 (2020).
Moteki, H. et al. Hearing loss caused by a mutation identified in a MELAS family with a coexisting mitochondrial 3243AG mutation. Ann. Oto. Rhinol. Laryn. 124, 177s–183s. https://doi.org/10.1177/0003489415575045 (2015).
Yan, D. et al. Mutation of the ATP-gated P2X2 receptor leads to progressive hearing loss and increased susceptibility to noise. Proc. Natl. Acad. Sci. 110, 2228–2233 (2013).
Liu, X. Z., Yan, D., Mittal, R., Ballard, M. E. & Feng, Y. Progressive dominant hearing loss (autosomal dominant deafness-41) and P2RX2 gene mutations: A phenotype-genotype study. Laryngoscope 130, 1657–1663 (2020).
Lee, J. et al. A database of 5305 healthy Korean individuals reveals genetic and clinical implications for an East Asian population. Exp. Mol. Med. 54, 1862–1871. https://doi.org/10.1038/s12276-022-00871-4 (2022).
Tadaka, S. et al. 3.5KJPNv2: An allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum Genome Var 6, 28. https://doi.org/10.1038/s41439-019-0059-5 (2019).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
Van der Auwera, G. A. et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Prot. Bioinf. 43, 11 (2013).
Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv, 201178 (2017).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164 (2010).
Abou Tayoun, A. N. et al. Improving hearing loss gene testing: A systematic review of gene evidence toward more efficient next-generation sequencing-based diagnostic testing and interpretation. Genet Med. 18, 545–553. https://doi.org/10.1038/gim.2015.141 (2016).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613. https://doi.org/10.1002/humu.23630 (2018).
Peng, J. et al. VIP-HL: Semi-automated ACMG/AMP variant interpretation platform for genetic hearing loss. Hum. Mutat. 42, 1567–1575 (2021).
Kirov, G. et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatr. 75, 378–385. https://doi.org/10.1016/j.biopsych.2013.07.022 (2014).
Minikel, E. V. et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 8, 322ra329. https://doi.org/10.1126/scitranslmed.aad5169 (2016).
Whiffin, N. et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 19, 1151–1158. https://doi.org/10.1038/gim.2017.26 (2017).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded (RS-2024-00400118 to H.Y.G.) and the Korea Health Industry Development Institute (KHIDI) grants (RS-2024-00438709 to H.Y.G. and RS-2024-00346485 to J.J).
Funding
Korea Health Industry Development Institute,RS-2024-00346485,RS-2024-00438709,National Research Foundation of Korea,RS-2024-00400118, RS-2025-02214844
Author information
Authors and Affiliations
Contributions
J.Y.C., J.J., and H.Y.G.: Conceptualization; S.Y.J., S.H.J., J.A.K., and S.J.K.: Formal analysis; J.J. and H.Y.G.: Funding acquisition; S.Y.J. and S.H.J.: Investigation; S.Y.J., J.A.K, and S.J.K.: Methodology; J.Y.C., J.J., and H.Y.G.: Resource; J.J. and H.Y.G.: Supervision; S.Y.J., S.H.J., J.A.K., and S.J.K.: Validation; S.Y.J., S.H.J., and H.Y.G.: Visualization; S.Y.J. S.H.J., and H.Y.G.: Writing—original draft; J.J. and H.Y.G.: Writing—review & editing. All the authors revised the manuscript and approved the final version for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Joo, S.Y., Jang, S.H., Kim, J.A. et al. Leveraging underrepresented population data improves interpretation of genetic variants associated with hearing loss. Sci Rep 15, 33693 (2025). https://doi.org/10.1038/s41598-025-18852-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18852-8
