
Abstract
Short chain fatty acids (SCFA) are key microbial metabolites that modulate intestinal homeostasis and may influence irritable bowel syndrome (IBS) pathophysiology. We aimed to assess microbial features associated with SCFA and determine if features varied across IBS subtypes and endophenotypes. We analyzed stool microbial metagenomes, stool SCFA, and measurable IBS traits (stool bile acids, colonic transit, stool form) in 41 patients with IBS (IBS with constipation [IBS-C] IBS with diarrhea [IBS-D]) and 17 healthy controls. We used partial canonical correspondence analyses (pCCA), conditioned on transit, to quantify microbe-SCFA associations across groups. We further compared gut microbiome-encoded potential for substrate utilization across groups and within a subset of participants selected by their stool characteristics as well as stool microbiomes of patients with and without clinical bile acid malabsorption (BAM). Microbe-SCFA associations differed across groups and revealed key taxa including Dorea sp. CAG:317 and Bifidobacterium pseudocatenulatum in IBS-D and Akkermansia muciniphila and Prevotella copri in IBS-C that that could underlie subtype-specific microbially-mediated mechanisms. The greatest number of microbe-SCFA associations were observed in IBS-D. Several SCFA-producing species demonstrated inverse correlations with SCFA. Fewer bacterial taxa were associated with acetate to butyrate ratios in IBS compared to health. In participants selected by stool form, we demonstrated differential abundances of microbial genes/pathways for SCFA metabolism and degradation of carbohydrates and mucin across groups. SCFA-producing taxa were reduced in IBS-D patients with BAM. Keystone taxa responsible for SCFA production differ by IBS subtype and traits. IBS microbiomes appear exhibit reduced functional redundancy. Differences in substrate preferences are also linked to bowel functions. Focusing on taxa that drive SCFA profiles and stool form may be a rational strategy for identifying relevant microbial targets in IBS.
Similar content being viewed by others

Introduction
Irritable bowel syndrome (IBS) is a burdensome disorder of gut-brain interaction with an estimated global prevalence rate of 5–10%1. Pathophysiological mechanisms of IBS include disturbances in motility or transit, altered intestinal secretion, impaired intestinal permeability, immune cell reactivity, visceral hypersensitivity, and dysregulated neural signaling and/or central processing2. Despite advancements in the understanding of IBS pathogenesis, diagnostic and therapeutic IBS biomarkers are limited.
Accumulating evidence suggests that the gastrointestinal (GI) microbiome is associated with risk of IBS and may also mediate many of the mechanisms that underlie symptoms including altered motility3,4, barrier dysfunction5, immune activation6,7, signaling along the brain-gut axis8, and visceral sensation9. Characterization of microbial composition in IBS has suggested decreased microbial diversity, reduced temporal stability, or changes in the relative abundance of specific bacteria in patients with IBS10,11. However, these findings have not been sufficiently consistent across studies to establish a clear microbial profile in IBS and gaps in our understanding of the functional microbiome persist. Integrating complementary strategies in the investigation of microbial metabolites will be crucial for gathering actionable insights into the impact of the microbiome in IBS.
Atypical profiles of microbial metabolites including luminal bile acids12,13 and short chain fatty acids (SCFA) have been described in some patients with IBS14,15. Bile acid malabsorption (BAM) is recognized as a mechanistic IBS subtype that can be assessed through several diagnostic methods including measurement of total or primary stool bile acids13. Studies have demonstrated BAM to be associated with physiological traits, symptoms, and quality of life16,17,18,19. Recently, researchers have examined microbial contributions to BAM in IBS to report alterations such as enrichment of Clostridia bacteria including C. scindens20, lower microbial alpha diversity, higher Firmicutes to Bacteroidetes ratio19, and presence of endoscopically visible biofilms correlating with overgrowth of Escherichia coli and Ruminococcus gnavus21.
SCFA are produced by anaerobic fermentation of dietary fibers and resistant starch that enter the colon and regulate intestinal homeostasis and physiology22. Compared to bile acids, the role of SCFA in IBS is less well understood. Studies of stool SCFA in IBS have yielded variable results, which may be related to the heterogeneity of the IBS patient populations and multiple pathways through which SCFA may modulate intestinal physiology. Therefore, while stool SCFA are unlikely to serve as categorical IBS biomarkers, they may provide critical insights into the pathophysiological mechanisms that underlie IBS symptoms or serve as a tool for identifying metabolically relevant microbial targets. Studies15,23 that have assessed stool SCFA in distinct IBS subgroups have reported more consistent associations of stool SCFA with IBS subtypes as well as correlations of stool SCFA with measurable IBS traits such as colonic transit, bowel functions, and bile acid excretion23,24,25. Despite these reports, the intercorrelation between SCFA and transit time complicates the assessment of whether SCFA profiles represent the metabolic capacity of the resident microbiome. It remains unclear if studying the relationships between the microbiome and excreted SCFA is clinically informative. Recent work26 has suggested that individual or keystone taxa, rather than complex ecological communities, could drive changes in SCFA output in response to dietary fiber. Therefore, strategies that isolate major microbial features (or keystone taxa) that shape luminal SCFA may serve as a rational method for selecting functionally relevant microbial targets in patients with IBS. To address these questions, we conducted an in-depth investigation of GI microbiome composition and function, stool SCFA, and IBS endophenotypes defined according to quantitative traits (transit, bile acids, bowel functions) in adults with and without IBS.
Results
Participant characteristics
Among 96 volunteers who underwent screening evaluation, 71 completed the study, and 58 participants (Fig. 1) with a mean [± SD] age = 35.5 (± 13.8) years and mean [± SD] BMI = 26.2 [± 7.5] kg/m2) were included in the final analysis after excluding those who ineligible (n = 16), lost to follow-up (n = 9), or and/or did not provide SCFA data (n = 13). Baseline clinical characteristics (Table 1) including macronutrient intake were not significantly different across groups (all p = ns). No participants reported a historical use of modified, exclusionary, or restrictive diets at the time of screening. Comparisons of quantitative traits demonstrated differences in total stool bile acids and transit between IBS-D and healthy volunteer (HV) participants and in total stool SCFA and stool acetate between IBS and HV Table 2; Figs. 2 and 3.

Consort Flow Diagram.

Stool Short Chain Fatty Acid Concentrations in Healthy Volunteers and Participants with Irritable Bowel Syndrome (IBS). Histograms are shown for total short chain fatty acids, acetate, butyrate, propionate, and acetate to butyrate ratios within clinical groups including healthy volunteers (HV), IBS with constipation (IBS-C), and IBS with diarrhea (IBS-D). Groups are denoted by color (HV = green, IBS-C = blue, IBS-D = red).

Stool Bile Acids in Healthy Volunteers and Participants with Irritable Bowel Syndrome (IBS). Histograms are shown for % primary bile acids (bile.acids) and total bile acids within clinical groups including healthy volunteers (HV), IBS with constipation (IBS-C), and IBS with diarrhea (IBS-D). Groups are denoted by color (HV = green, IBS-C = blue, IBS-D = red).
Stool microbiome composition differs between IBS and health and between IBS subtypes
Shotgun metagenomic sequencing of stool samples was undertaken to obtain total of 3.1 Gb of sequence data after removal of contaminants with an average of 37.6 million paired-end reads per sample (deposited into NCBI with accession number PRJNA1023929). Taxonomic classification identified 461 taxa at species level classification. Comparisons of microbial metagenomes based on Bray-Curtis Dissimilarity revealed significant divergence (Supplemental Fig. 1) of community distance between groups in both unadjusted analyses (p = 0.01) and after adjusting for age, BMI, and dietary variables selected by Random Forest algorithm (p = 0.003). We compared abundances of individual taxa across groups using the Kruskal-Wallis test to identify 18 unique and differentially abundant taxa. Highest mean relative abundances of 14 taxa (Supplemental Table 1) including Dorea sp. CAG:317, Blautia sp. CAG:257, Ruminococcus gnavus, and Proteobacteria bacterium CAG:139 were observed in IBS-D, which have previously been linked to mechanisms such as BAM20,21 and serotonin biosynthesis in patients with IBS-D27. Lawsonibacter asaccharolyticus abundance was highest in IBS-C and Firmicutes bacterium CAG:83 abundance was highest in HV.
We followed unadjusted analyses with pairwise comparisons of ALR-transformed taxa abundances using covariate-adjusted GLM, focusing on high abundant species, to demonstrate significant differences in 17 pairwise comparisons including 12 unique species (Fig. 4) of which 11 exhibited differential abundances of ≥ 3-fold. Among these taxa, we observed significantly higher abundances of Dorea sp. CAG:317 and Bifidobacterium pseudocatenulatum in IBS-D compared to IBS-C or HV and higher abundances of Blautia sp. CAG:257, and Proteobacteria bacterium CAG:139 in IBS-D compared to HV. We found significantly higher abundances of Clostridium sp. CAG:58 and lower abundances of Firmicutes bacterium CAG:83 in both IBS-D and IBS-C relative to HV, respectively. Akkermansia muciniphila and Prevotella copri were increased in IBS-C compared to HV. Differences of ≥ 3-fold in relative abundances among high abundant bacteria were also observed in 151 pairwise comparisons (Supplemental Table 2) but were not statistically significant after adjustment for covariates.

Bacterial Taxa with Differential Abundance of ≥ 3-fold Between Groups. Data show mean percent abundances (standard deviation) of significantly different bacterial taxa with ≥ 3-fold differences in pairwise comparisons of clinical cohorts.
Microbe-SCFA associations differ between IBS subtypes and HV
We conducted pCCA on microbiome and SCFA data, conditioned on transit time, to quantify relationships between taxa abundances and stool SCFA in the overall cohort and within clinical groups. In the overall cohort, we observed a significant correlation between microbiome abundance matrix and SCFA concentration matrix (p = 0.033, R2 = 0.095). Relationships between microbiome abundances and SCFA concentrations within clinical groups (Supplemental Fig. 2) were more pronounced within IBS-D (p = 0.015) and less pronounced in HV and IBS-C (p = ns). In the overall cohort, the largest positive associations with total SCFA were observed with Bacteroides plebeius, Prevotella sp. CAG:1031, and Bifidobacterium pseudocatenulatum.
Distinct taxa correlate with individual SCFA across clinical subgroups
Keeping in mind that SCFA effects may depend on SCFA type, we ranked bacterial taxa according to their projection scores (Supplemental Tables 3–6) for individual SCFAs to assess the pattern of associations between bacterial species and SCFA profiles. A higher projection score for an individual SCFA indicates a greater degree of association with that specific SCFA. Bacteria with projection scores less than 0.5 were excluded from the subsequent analysis. In general, the degree and direction of correlations of individual taxa with acetate, butyrate, and propionate differed according to clinical group (Fig. 5). The greatest number of microbe-SCFA associations were observed in IBS-D, as evidenced by the larger number of bacteria remaining in the ranked list after filtering. Most microbe-SCFA patterns differed between IBS-D and IBS-C and only a few bacterial species including Ruminococcus torques, Coprococcus comes, Clostridium sp. CAG:299, Bacteroides eggerthii, and Adlercreutzia equolifaciens demonstrated consistent associations across both IBS subtypes with acetate and butyrate.

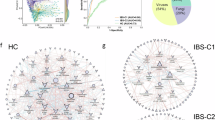
Correlations of bacterial taxa with short chain fatty acids (SCFA) based on partial canonical correspondence analysis. Panels show associations between bacterial taxa and specific SCFA ([a] acetate, [b] acetate to butyrate ratio, [c] butyrate, and [d] and propionate) within cohorts. The strength of the correlation is represented projections > 0.5 with purple indicating negative correlations and red indicating positive correlations.
In IBS-D, both positive and negative correlations with stool acetate were observed with multiple taxa. Largest negative projections were observed with several bacteria that have previously linked to SCFA production (B. plebeius, Roseburia hominis)28,29, dietary polysaccharides and protein utilizers (Firmicutes bacterium CAG:110)30, starch or fructooligosaccharide degradation (Bifidobacterium adolescentis), fiber fermentation (e.g. Clostridium leptum31, β-fructan utilizers (e.g. Roseburia inulinivorans)31. Positive projections were observed with Dialister sp. CAG 357, B. pseudocatenulatum, Alistipes species, Prevotella sp. CAG:1031, R. toques, and R. gnavus. In IBS-C, largest negative projections for acetate were observed between abundances of Dorea sp. CAG:317, Lachnospira pectinoschiza, and Prevotella sp. CAG:1031 while the largest positive projections were observed with Streptococcus salivarus, Bacteroides fragilis, and R. torques. In HV, largest negative projections for acetate were observed with B. plebeius, Dialister sp. CAG:357, Bacteroides dorei, and A. muciniphila. The largest positive projection for acetate among HV was observed with Ruminococcus lactaris.
Analysis of microbe-butyrate associations in IBS-D demonstrated negative projection scores for butyrate with several bacteria including Ruminococcaceae bacterium D5, Firmicutes bacterium CAG:110, C. leptum, R. hominis, R. inulinivorans, and L. asaccharolyticus. The largest positive projection scores for butyrate were observed with Alistipes species, Prevotella sp. CAG:1031, and R. torques. In IBS-C and HV, patterns of microbe-butyrate associations largely mirrored the patterns of microbe-acetate associations observed within the respective clinical groups. In addition, a positive relationship between butyrate and Bacteroides thetaiotaomicron, a well-known human commensal32,33 that exhibits the capacity to digest a broad array of polysaccharides and host glycans, was also observed in IBS-C.
For propionate, most microbes including Ruminococcaceae bacterium D5, Firmicutes bacterium CAG:110, C. leptum, L. asaccharolyticus, Ruthenibacterium lactatiformans and Bacteroides massiliensis exhibited negative associations in IBS-D with only a few exceptions including S. salivarus, for which the largest positive correlation was observed. In contrast, both positive and negative projections for stool propionate were observed in IBS-C; the largest positive scores were observed for Paraprevotella xylaniphila, Phascolarctobacterium succinatutens, and P. copri. Among HV, both positive and negative associations with propionate were observed for multiple bacterial taxa.
Network-based validation of microbe–metabolite associations
The results of Bayesian network analyses (Supplemental Fig. 3) revealed consistent patterns with the projection score analysis. Notably, many taxa with high projection scores for individual SCFAs (e.g., R. torques, R. hominis, R. inulinivorans, C. leptum, B. plebeius, Prevotella sp. CAG:1031, etc.) were also found to be directly or indirectly associated with SCFAs in the Bayesian networks.
Lower number of bacterial species associated with acetate to butyrate ratios in IBS
Since interconversion of SCFA from acetate to butyrate may represent a major route of butyrate formation within the gastrointestinal tract34, we examined bacterial relationships with acetate to butyrate ratios to identify top taxa associated the metabolic processes that drive SCFA output. The largest positive associations were observed with Prevotella sp. 885, Firmicutes bacterium CAG:83, P. xylaniphila in IBS-D, suggesting that these species were associated with shift toward decreased butyrate production. Unlike the patterns observed with individual SCFA, analysis acetate to butyrate ratios demonstrated that the number of ranked bacteria were substantially lower in IBS-D or IBS-C compared to controls (Fig. 5), suggesting the possibility of reduced functional redundancy for butyrate formation in IBS.
Reduced SCFA-producing microbes and bile acid malabsorption (BAM)
Stool microbial metagenomes were analyzed in 22 IBS-D patients with (n = 8) and without (n = 14) clinical BAM according to previously validated diagnostic cutoff values12,13. Comparisons of beta diversity based on Euclidian dissimilarity of ALR transformed abundance data showed significant dissimilarity (p = 0.039; R2 = 0.061) between patients with and without BAM (Supplemental Fig. 4). Significant differences in six high abundant species were observed between groups (Supplemental Table 7), including L. asaccharolyticus R. inulinivorans, L. pectinoschiza, and F. saccharivorans which were also associated with stool SCFA, but not bile acids, in IBS-D. These four species with known SCFA-producing capacity were negatively correlated with BAM.
Functional potential for carbohydrate degradation, SCFA metabolism, and mucin degradation
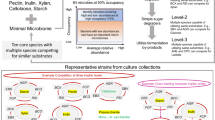
Examination of microbial gene families and pathways yielded 949,223 total gene families within the metagenome data set including 4,042 named KO terms. We applied a gut metabolic module (GMM) framework to analyze modules related to carbohydrate degradation, SCFA production or metabolism including bacterial cross-feeding pathways, mucin degradation, and one module related to serine degradation based on significant species-based predictors. Abundances of KO identifiers within modules for lactose degradation, (p = 0.0026), serine degradation (p = 0.019), and propionate production (p = 0.026) were significantly enriched in IBS-C compared to controls (Supplemental Table 8). Relative abundance of a KO identifier within a galactose degradation module was significantly reduced in IBS-D (p = 0.033) compared to controls. In the collective cohort, we applied the GMM framework to analyze the relationships between metagenomically-encoded functions and stool SCFA. We identified 23, 23, 15, and 16 KO identifiers from relevant GMMs that were significantly associated with total SCFA, acetate, butyrate, and propionate, respectively (Supplemental Tables 9–12). Within a representative subset (n = 6 controls, n = 5 IBS-D, n = 5 IBS-C) selected by stool form characteristics, functional metagenomic analyses demonstrated that microbial genes/pathways associated with SCFA production/metabolism and degradation of carbohydrates/mucin were differentially associated with clinical group (Fig. 6).

Permission through Kanehisa Laboratories was not requested as Fig. 6was independently generated and we did not use any images or diagrams from Kanehisa Laboratories.
Variations in Microbial Substrate Utilization in Healthy Volunteers and Participants with IBS. Figure shows significant differences in functional potential for carbohydrate degradation, short chain fatty acid production or metabolism, and mucin degradation across clinical groups in representative subset selected by stool form characteristics. Groups are denoted by color (HV = blue, IBS-C = red, IBS-D = gray). .
Discussion
It has been proposed that changes in intestinal SCFAs in some patients with IBS are caused by shifts in microbial composition and function to drive and maintain symptoms. We hypothesized that specific microbiome features correlate with SCFA output and that these relationships vary across IBS subtypes and endophenotypes. Using a dual-omics approach, we found that microbiota-SCFA relationships differ between IBS subtypes and endophenotypes, and between patients with IBS and HV. The focus on paired microbe-SCFA data was grounded in the known biological relevance of SCFA in GI physiology. Our findings suggest that key taxa may shape the microbial metabolome, contribute to IBS mechanisms, and influence bowel functions through their functional capacities.
While many studies have reported compositional changes11 in IBS, consistent patterns are elusive. In our study, we observed differences in overall microbial community composition across groups with significant changes being most apparent in IBS-D. These alterations were predominantly characterized by higher abundances of several bacterial species including Blautia sp. CAG:257, R. gnavus, Dorea sp. CAG:317, and Proteobacteria bacterium CAG:139. R. gnavus, has previously been implicated in IBS-D pathogenesis through serotonin biosynthesis27, biofilm formation and increased stool bile acid excretion21, production of proinflammatory polysaccharides35, and mucin degradation36. Interestingly, the relationship between R. gnavus and IBS lost its significant in adjusted analyses due to a positive association between R. gnavus and BMI, which may suggest a role for R. gnavus in explaining the connection between IBS-D or chronic diarrhea with metabolic syndrome and obesity-related disorders37. Associations of IBS-D with other taxa including Dorea sp. CAG:317, Blautia sp. CAG:257, and Proteobacteria bacterium CAG:139 remained and a new association between B. pseudocatenulatum and IBS-D emerged.
In our analyses of microbe-SCFA relationships, largest contributions to SCFA in the overall cohort were observed with B. plebeius, Prevotella sp. CAG:1031, and B. pseudocatenulatum, indicating their central role in microbial metabolism. These taxa possess adaptations for dietary carbohydrate fermentation, supporting their metabolic relevance38,39,40,41. In IBS-D, microbe-SCFA associations were most pronounced in IBS-D and fewer microbes were linked to acetate and butyrate ratios. These observations may indicate that microbial contributions to SCFA profiles are particularly important in IBS-D and could also imply a greater degree of microbial specialization and reduced functional redundancy. Previously, Jacobs et al.42 reported significant increases in transcript abundances for fructooligosaccharide and polyol utilization and upregulation of transcripts for fructose and glucan metabolism as well as the succinate pathway for carbohydrate fermentation in IBS. Comparisons between IBS-D and IBS-C demonstrated differences in microbial metabolism with upregulation of multiple metabolic pathways including transcripts for fructose, mannose, and polyol metabolism in IBS-D. Together, our findings suggest that differences microbial metabolism may underlie variations in clinical symptoms or pathophysiological mechanisms across IBS subtypes and that altered microbial metabolism could be particularly important in IBS-D. Our findings may further explain the higher prevalence of mixed- and diarrhea-predominant symptoms in post-infection IBS43,44 as well as the limited evidence for efficacy of antimicrobial treatments such as rifaximin in IBS-C45 relative to IBS-D46,47.
Dorea sp. CAG:317 and B. pseudocatenulatum were identified as top ranked bacteria associated with acetate to butyrate ratio and acetate concentrations. Mucin degradation has been described among several Dorea species and the genus Dorea belongs to the Lachnospiraceae family, a major producer of SCFA48, Notably, Wang et al.49 recently demonstrated that while xylan supplementation following fiber deprivation alleviated gut dysbiosis by promoting B. pseudocatenulatum, these changes were associated with lower community diversity. Therefore, Dorea sp. CAG:317 and B. pseudocatenulatum may represent keystone taxa linked to altered microbial metabolism or persistent impairment in community recovery among patients with IBS-D. Only two bacterial species, A. muciniphila and P. copri were significantly increased in IBS-C. P. copri was further identified as top ranked bacteria that was positively associated with propionate and negatively associated with acetate to butyrate ratio in IBS-C. Previous studies have reported a positive association between mucosal P. copri abundance and abdominal pain in patients with IBS without prior history of infection50. Others have demonstrated that IBS patients with high levels of propionate present with worse gastrointestinal symptoms, quality of life, and negative emotions15. P. copri is a prominent gut commensal that contributes to propionate production via the succinate pathway51 and has been linked to both beneficial and harmful effects on human health. In another study by Jiang et al., P. copri colonization with high fiber diets was associated with proinflammatory effects and Akkermansia expansion in patients with rheumatoid arthritis52. Together, these observations may indicate P. copri–driven propionate production, particularly in the context of increased Akkermansia could promote pathophysiological mechanisms (e.g., immune activation) that drive symptom burden in in IBS-C.
In addition to identify major microbial features associated with individual SCFA, we made several important observations related to the pattern or direction of observed microbe-SCFA relationships. Interestingly, negative correlations were observed in the IBS-D subgroup between several SCFA-producing species including R. hominis, B. adolescentis, R. inulinivorans and individual SCFA. Findings may imply disrupted cross-feeding mechanisms, modifications in substrate utilization, competitive inhibition, or other context-specific changes leading to altered microbial metabolism in IBS. R. inulinivorans is a known fiber fermenter31 that also possesses the ability to forage mucin through a metabolic interplay with other members of the intestinal microbiota53 and is known for net acetate uptake51, which may explain its correlation with reduced SCFA. Upon analyzing relationships of bacterial species with acetate to butyrate ratios, which we studied as a marker for overall SCFA production and luminal SCFA availability54, we identified fewer top ranked bacteria in IBS compared to healthy controls, suggesting a reduction in overall functional redundancy of the IBS microbiome. Future work should investigate the effects of bacterial cross-feeding, microbial conversions, and interactions of the colonic microbiome with diet- and host-derived substrates that may destabilize the microbiota-mucosal interface to expose vulnerabilities and that drive IBS pathogenesis.
A few bacterial species were associated with SCFA in both IBS subtypes including R. torques, C. comes, Clostridium sp. CAG:299, B. eggerthii, and Adlercreutzia equolifaciens which may suggest these bacterial species represent shared microbial features across IBS subtypes. R. torques is a mucin-degrading bacteria that has previously been associated with IBS55 as well as depression56. Similarly, recent studies have demonstrated associations between C. comes57 and B. eggerthii58 depression and post-traumatic stress disorder, which have both been described as comorbid conditions in IBS. These data could suggest that some microbial features are linked to common pathophysiologic mechanisms involving the microbiota-gut-brain axis that are shared across IBS subtypes and that their effects may be mediated by microbially-derived SCFA.
In our comparisons across IBS-D patients by with and without BAM, we observed BAM to be associated with reduced abundances of several SCFA-producing species. Similar observations have been made in studies linking biofilm formation to bile acid accumulation and reduced SCFA-producing bacteria21. Results suggest that BAM exerts indirect effects on the intestinal metabolome and may associated with a decline in SCFA-producing capacity. We also assessed of functional variations in the GI microbiome to find differential abundances of KO identifiers for metabolic processes related to degradation of carbohydrates and serine as well as SCFA production. In a subset of participants selected by stool form characteristics, we found differences in substrate degradation potential and fermentative capacity across groups including increased capacity for starch degradation in controls, alterations in distinct mucin-degrading functions in IBS-D vs. IBS-C, and reduced pectin degrading potential in IBS-C. Results suggest that abnormal stool form in IBS is influenced by microbiota-encoded substrate preferences.
Collectively, our findings indicate there are distinct changes in microbiome composition may underlie clinical heterogeneity in IBS. We demonstrate that the gut microbiota in IBS-D is characterized by increased metabolic activity, reduced functional redundancy, and persistently reduced diversity. Several taxa including R. gnavus, Dorea sp. CAG:317 and B. pseudocatenulatum emerged as the leading candidates for further study as potential microbial targets in IBS-D. A few unique microbial features including P. copri-associated propionate production and Akkermansia expansion may play a crucial role in IBS-C. Meanwhile, microbial features that are common between subtypes may offer insights into shared pathophysiological mechanisms involving the microbiota-gut-brain axis in IBS. Identifying the major microbial features (taxa or functions) that drive metabolic output may be crucial in defining rational approaches to modulate the microbiome in IBS.
Despite study strengths, we recognize the limitations of this work including cross-sectional sampling, which overlooks the fluctuating nature of the microbiome. To address this, we assessed relationships between microbial abundances and quantifiable IBS endophenotypes that were defined at the time of specimen collection while accounting for baseline covariates. All participants were instructed to consume a 4-day high fat diet while otherwise maintaining their usual diet. Previously, we examined the effect of both habitual diet and real-time intake on excreted stool SCFA to find that that modest variations in macronutrient intake including polysaccharides did not exert substantial effects on excreted SCFA25,59. We enrolled patients with opposing IBS phenotypes from both the university clinics and the surrounding communities to increase our ability to detect differences between clinical cohorts and limit referral bias. We also acknowledge that causal microbial mechanisms cannot be determined through the current work. However, we applied a dual-omics approach to investigate biologically relevant changes in intestinal microbiome composition and function. We cannot discount the possibility we may have missed significant taxa due to the moderate sample size and a risk for Type I errors as we did not adjust for false discovery rate in our analyses of taxonomic differences. Moreover, the choice of metagenomic classification tool may influence taxonomic profiling60. Hence, results should be considered as hypothesis-generating. However, our analytic approach was based on endpoints that were determined a priori and we applied a conservative strategy by focusing on taxa that were associated with SCFA while accounting for transit. We further limited analyses to high abundant taxa and to species that exhibited > 3-fold change in abundance while focusing on the biological plausibility of our results. MetaPhlAn is a widely used metagenomic classification tool that relies on clade-specific marker genes is well-suited for identifying species-level patterns in human samples. A complementary Bayesian Network Analysis was also conducted to better capture microbial interactions and shared influences on SCFA profiles. Importantly, many of the taxa identified via projection scores were noted to occupy structurally important positions in the broader microbiome-metabolite-transit interaction network, supporting the biological relevance of our findings. Finally, we compared IBS subtypes, but did not evaluate symptom severity with tools such as the IBS severity scoring system61, which others have shown may be correlated with microbiome composition3. Findings from this study should be validated in larger, longitudinal cohorts and investigate other aspects of IBS phenotypes such as symptom severity, but still provide a novel framework that could be used to uncover key microbial interactions and targets in IBS.
Conclusions
In conclusion, main findings from this study highlight microbiota-SCFA patterns vary across clinical IBS phenotypes and endophenotypes. Prominent shifts in microbial composition are observed in IBS-D. These changes appear to affect metabolic capacity, substrate preferences, and metabolite profiles. In addition, altered microbial substrate uptake may impact stool form or bowel function in IBS. Importantly, we identify specific taxa—such as R. gnavus, Dorea spp., and B. pseudocatenulatum—that may serve as biomarkers or therapeutic targets due to their strong association with SCFA profiles. Our findings support the utility of a paired microbe-metabolite approach as a viable strategy for identifying functionally relevant microbial features in heterogenous IBS patient populations.
Materials and methods
Participant recruitment and study design
The study was approved by the Indiana University Institutional Review Board and the protocol registered within ClinicalTrials.gov (NCT02981888). The study was conducted in accordance with the principles of the Declaration of Helsinki and all patients provided written informed consent before enrollment. The study was designed as an observational investigation of stool SCFA, stool bile acids, colonic transit, and stool microbiota in adults with and without IBS. We enrolled adults ages 18–65 years of age through the Indiana University Gastroenterology Clinics, the Indiana Clinical and Translational Research Institute Research Registry, and from the local community. We included individuals with IBS with diarrhea (IBS-D) or IBS with constipation (IBS-C) according to Rome IV criteria62 and healthy controls with no prior history of GI diseases or symptoms. Detailed eligibility criteria are available in the Supplemental Methods.
Data collection
Study eligibility, medications, medical history, and baseline diet using a food frequency questionnaire63 were assessed during a screening visit with a study physician. Over a two-week period, data were collected on bowel functions using a standardized bowel pattern diary including the Bristol stool form scale64. All participants were instructed to maintain their habitual diets and submitted a 48-hour stool collection collected during the last 2 days of a 4-day 100 g fat diet, consistent with clinically validated methods for identifying BAM. Specimens were refrigerated during the collection period, returned to the research team on ice, and stored at −80οC.
Colonic transit by radiopaque markers
Participants underwent assessment of colonic transit time with a previously validated and optimized method using radiopaque markers65.
Stool SCFA and bile acids
Frozen aliquots of stool were shipped to the Metabolite Profiling Facility at Purdue University to measure total and individual SCFA concentrations per mg of dry weight by liquid chromatography-mass spectrometry (LC-MS) using published methods66 and to the Mayo Clinic Department of Laboratory Medicine and Pathology to measure total and primary stool bile acid levels by high-performance LC-MS through a commercially available, CLIA-approved assay16,67,68. Individual SCFA of interest included acetate, butyrate, and propionate as these represent the predominant SCFA produced in humans69.
DNA extraction, purification, and sequencing
Genomic DNA was isolated from stool using the QIAmp® PowerFecal® DNA kit (QIAGEN Inc., Germantown, MD, USA). DNA quality and concentration were on a Qubit fluorometer. Purified DNA underwent library preparation (Nextera XT, Illumina) and paired-end (2 × 150 bp) sequencing using the NovaSeq v1.5 SP (Illumina, San Diego, CA, USA) to target a sequencing depth of 40 M sequences per sample.
Metagenomic data analysis
Metagenomic sequencing reads were quality filtered and processed for taxonomic profiling using MetaPhlAn3. Additive log-ratio (ALR) transformation was applied to analyze differential taxonomic abundances. Based on the identified microbial taxa from each sample, β-diversity indices (e.g., Bray-Curtis dissimilarity) were calculated using the R packages phyloseq and vegan70,71. Functional profiling was conducted using the HUMAnN3 pipeline annotated by KEGG (Kyoto Encyclopedia of Genes and Genomes) orthogroups (KO)72,73,74.
Statistical considerations
We summarized major endpoints of interest: (1) stool microbiome composition, (2) stool SCFA concentrations (total, acetate, propionate, butyrate), total and percent primary stool bile acids, and (3) colonic transit time (overall and segmental). Our primary objective was to quantify the association between microbiome composition and SCFA (microbe-SCFA associations) across clinical groups after controlling for transit and to account for the mechanistic heterogeneity that underlies clinical IBS populations.
For overall community composition, we compared microbial community dissimilarity using the PERMANOVA test across groups (IBS-C, IBS-D, controls) with and without adjusting for covariates (age, BMI, dietary variables) selected by the Random Forest algorithm. Dietary variables suggested by the machine-learning technique included starch, total energy, and protein. Significant associations of community distance by Bray-Curtis Dissimilarity with group were observed in both diet-adjusted and unadjusted analyses. We compared microbial taxa abundances between groups and analyzed associations of taxa abundances with IBS endophenotypes/traits (stool SCFA, stool bile acids, and transit) for the collective cohort and within groups. Associations of microbiome composition with group were assessed using general linear regression models (GLM) adjusted for covariates including age, sex, and BMI. Only high abundant species (relative abundance ≥ 0.1%) that were prevalent in ≥ 2 specimens within ≥ 1 clinical group were considered. Partial Canonical Correspondence Analysis (pCCA) conditioned on transit time was employed to Quantify the relationship between the microbiome and stool SCFA in all participants and within clinical groups, using the vegan package in R. Conditioning on transit time accounted for variation in microbial and metabolite profiles attributable solely to differences in gastrointestinal transit. The significance of the model was assessed through a permutation test with 999 iterations. To identify bacteria whose abundances exhibited the greatest degree of association with the stool SCFA concentrations, we focused on those bacteria scattered along the direction of specific SCFA axis in the biplot, within a 60-degree angle centered around the SCFA axis in both positive and negative directions. The strength of association between the bacteria and the SCFA was ranked by projecting of the bacteria species scores onto each SCFA axis in the biplot. Bacteria with projection scores less than 0.5 were excluded from the subsequent analysis. To complement the projection score approach and explore the complexity of microbiome–metabolite–transit interactions, we conducted a Bayesian Network Analysis with R package bnlearn. The network structure was inferred using a bootstrapped ensemble of 500 iterations and only edges that appeared in at least 50% of bootstraps were retained. All associations were included (i.e., partial correlation coefficient > 0).
For exploratory analyses, we analyzed multivariable associations of microbiome composition with clinical BAM among individuals with IBS-D. Relative abundances were used to examine the associations of gene family/pathway abundances across clinical groups. A manually curated gut-metabolic analysis framework75 was applied to examine KO identifiers associated with carbohydrate degradation, SCFA production or metabolism, and mucin degradation using GLM adjusting for covariates. As stool form76 has been identified as a strong source of human microbiota variation and closely linked to transit77,78, we further explored associations of functional potential across groups in a subset of participants who were selected based on stool form features (i.e. those individuals best representing the stool types within their respective clinical group according to bowel diary data) using the Kruskal-Wallis test. For example, we chose the IBS-D and IBS-C participants with the loosest and firmest stool types, respectively, and controls with consistently normal stool types. For all endpoints, missing values were excluded from the analysis for that endpoint.
Data availability
Microbiome sequencing reads are deposited into NCBI with accession number PRJNA1023929. All relevant data generated during the study and the R codes used to conduct the analyses for this study are included in this published article (and its supplementary information files) or are available for download from GitHub via the following link: https://github.com/xianggao2006/IBS (accessed on 1 May 2025).
Abbreviations
- ALR:
-
Additive log-ratio
- BAM:
-
Bile acid malabsorption
- CTT:
-
Colonic transit time
- gDNA:
-
Genomic DNA
- GI:
-
Gastrointestinal
- GLM:
-
Generalized linear model
- GMM:
-
Gut metabolic module
- HV:
-
Healthy volunteer
- IBS:
-
Irritable bowel syndrome
- IBS-D:
-
IBS with diarrhea
- IBS-C:
-
IBS with constipation
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomics
- KO:
-
KEGG Orthogroup
- LC-MS:
-
Liquid chromatography-mass spectrometry
- SCFA:
-
Short chain fatty acids
References
Sperber, A. D. et al. Worldwide Prevalence and Burden of Functional Gastrointestinal Disorders, Results of Rome Foundation Global Study. Gastroenterology 160, 99–114 e113 (2021). https://doi.org/10.1053/j.gastro.2020.04.014
Ford, A. C., Sperber, A. D., Corsetti, M. & Camilleri, M. Irritable bowel syndrome. Lancet 396, 1675–1688. https://doi.org/10.1016/S0140-6736(20)31548-8 (2020).
Tap, J. et al. Identification of an Intestinal Microbiota Signature Associated With Severity of Irritable Bowel Syndrome. Gastroenterology 152, 111–123 e118 (2017). https://doi.org/10.1053/j.gastro.2016.09.049
Anitha, M., Vijay-Kumar, M., Sitaraman, S. V., Gewirtz, A. T. & Srinivasan, S. Gut microbial products regulate murine gastrointestinal motility via Toll-like receptor 4 signaling. Gastroenterology 143, 1006–1016 e1004 (2012). https://doi.org/10.1053/j.gastro.2012.06.034
Heitkemper, M. M. et al. Stool and urine trefoil factor 3 levels: associations with symptoms, intestinal permeability, and microbial diversity in irritable bowel syndrome. Benef Microbes. 9, 345–355. https://doi.org/10.3920/BM2017.0059 (2018).
Sundin, J., Rangel, I., Repsilber, D. & Brummer, R. J. Cytokine response after stimulation with key commensal bacteria differ in Post-Infectious irritable bowel syndrome (PI-IBS) patients compared to healthy controls. PLoS One. 10, e0134836. https://doi.org/10.1371/journal.pone.0134836 (2015).
McCracken, V. J. & Lorenz, R. G. The Gastrointestinal ecosystem: a precarious alliance among epithelium, immunity and microbiota. Cell. Microbiol. 3, 1–11. https://doi.org/10.1046/j.1462-5822.2001.00090.x (2001).
Liu, Y. et al. Similar fecal microbiota signatures in patients with Diarrhea-Predominant irritable bowel syndrome and patients with depression. Clin. Gastroenterol. Hepatol. 14, 1602–1611. https://doi.org/10.1016/j.cgh.2016.05.033 (2016). e1605.
Crouzet, L. et al. The hypersensitivity to colonic distension of IBS patients can be transferred to rats through their fecal microbiota. Neurogastroenterol Motil. 25, e272–282. https://doi.org/10.1111/nmo.12103 (2013).
Mars, R. A. T., Frith, M. & Kashyap, P. C. Functional Gastrointestinal disorders and the Microbiome-What is the best strategy for moving Microbiome-based therapies for functional Gastrointestinal disorders into the clinic?? Gastroenterology 160, 538–555. https://doi.org/10.1053/j.gastro.2020.10.058 (2021).
Pittayanon, R. et al. Gut microbiota in patients with irritable bowel Syndrome-A systematic review. Gastroenterology 157, 97–108. https://doi.org/10.1053/j.gastro.2019.03.049 (2019).
Shin, A. et al. Bowel functions, fecal unconjugated primary and secondary bile acids, and colonic transit in patients with irritable bowel syndrome. Clin. Gastroenterol. Hepatol. 11, 1270–1275. https://doi.org/10.1016/j.cgh.2013.04.020 (2013). e1271.
Vijayvargiya, P. et al. Bile and fat excretion are biomarkers of clinically significant diarrhoea and constipation in irritable bowel syndrome. Aliment. Pharmacol. Ther. 49, 744–758. https://doi.org/10.1111/apt.15106 (2019).
Mars, R. A. T. et al. Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome. Cell 182, 1460–1473 e1417 (2020). https://doi.org/10.1016/j.cell.2020.08.007
Tana, C. et al. Altered profiles of intestinal microbiota and organic acids May be the origin of symptoms in irritable bowel syndrome. Neurogastroenterol Motil. 22, 512–519. https://doi.org/10.1111/j.1365-2982.2009.01427.x (2010).
Vijayvargiya, P. et al. Analysis of Fecal Primary Bile Acids Detects Increased Stool Weight and Colonic Transit in Patients With Chronic Functional Diarrhea. Clin Gastroenterol Hepatol 17, 922–929 e922 (2019). https://doi.org/10.1016/j.cgh.2018.05.050
BouSaba, J. et al. Impact of Bile Acid Diarrhea in Patients With Diarrhea-Predominant Irritable Bowel Syndrome on Symptoms and Quality of Life. Clin Gastroenterol Hepatol 20, 2083–2090 e2081 (2022). https://doi.org/10.1016/j.cgh.2021.11.035
BouSaba, J. et al. Effect of rapid colonic transit on stool Microbiome and short-chain fatty acids in diarrhoea-predominant irritable bowel syndrome. Gut https://doi.org/10.1136/gutjnl-2022-329359 (2023).
Camilleri, M. et al. Comparison of biochemical, microbial and mucosal mRNA expression in bile acid diarrhoea and irritable bowel syndrome with diarrhoea. Gut 72, 54–65. https://doi.org/10.1136/gutjnl-2022-327471 (2023).
Zhao, L. et al. A Clostridia-rich microbiota enhances bile acid excretion in diarrhea-predominant irritable bowel syndrome. J. Clin. Invest. 130, 438–450. https://doi.org/10.1172/JCI130976 (2020).
Baumgartner, M. et al. Mucosal Biofilms Are an Endoscopic Feature of Irritable Bowel Syndrome and Ulcerative Colitis. Gastroenterology 161, 1245–1256 e1220 (2021). https://doi.org/10.1053/j.gastro.2021.06.024
Silva, Y. P., Bernardi, A. & Frozza, R. L. The role of Short-Chain fatty acids from gut microbiota in gut-Brain communication. Front. Endocrinol. (Lausanne). 11, 25. https://doi.org/10.3389/fendo.2020.00025 (2020).
Ringel-Kulka, T. et al. Altered colonic bacterial fermentation as a potential pathophysiological factor in irritable bowel syndrome. Am. J. Gastroenterol. 110, 1339–1346. https://doi.org/10.1038/ajg.2015.220 (2015).
Gargari, G. et al. Fecal clostridiales distribution and short-chain fatty acids reflect bowel habits in irritable bowel syndrome. Environ. Microbiol. 20, 3201–3213. https://doi.org/10.1111/1462-2920.14271 (2018).
Waseem, M. R. et al. Associations of fecal short chain fatty acids with colonic transit, fecal bile acid, and food intake in irritable bowel syndrome. Clin. Transl Gastroenterol. 14, e00541. https://doi.org/10.14309/ctg.0000000000000541 (2023).
Deehan, E. C. et al. Precision Microbiome Modulation with Discrete Dietary Fiber Structures Directs Short-Chain Fatty Acid Production. Cell Host Microbe 27, 389–404 e386 (2020). https://doi.org/10.1016/j.chom.2020.01.006
Zhai, L. et al. Ruminococcus gnavus plays a pathogenic role in diarrhea-predominant irritable bowel syndrome by increasing serotonin biosynthesis. Cell Host Microbe 31, 33–44 e35 (2023). https://doi.org/10.1016/j.chom.2022.11.006
Yun, E. J. et al. Metabolic and enzymatic Elucidation of cooperative degradation of red seaweed agarose by two human gut bacteria. Sci. Rep. 11, 13955. https://doi.org/10.1038/s41598-021-92872-y (2021).
Machiels, K. et al. A decrease of the butyrate-producing species roseburia hominis and Faecalibacterium Prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283. https://doi.org/10.1136/gutjnl-2013-304833 (2014).
Quan, J. et al. Metagenomic characterization of intestinal regions in pigs with contrasting feed efficiency. Front. Microbiol. 11, 32. https://doi.org/10.3389/fmicb.2020.00032 (2020).
Armstrong, H. K. et al. Unfermented beta-fructan fibers fuel inflammation in select inflammatory bowel disease patients. Gastroenterology 164, 228–240. https://doi.org/10.1053/j.gastro.2022.09.034 (2023).
Martens, E. C., Chiang, H. C. & Gordon, J. I. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell. Host Microbe. 4, 447–457. https://doi.org/10.1016/j.chom.2008.09.007 (2008).
Porter, N. T., Luis, A. S. & Martens, E. C. Bacteroides Thetaiotaomicron. Trends Microbiol. 26, 966–967. https://doi.org/10.1016/j.tim.2018.08.005 (2018).
Shetty, S. A., Boeren, S., Bui, T. P. N., Smidt, H. & de Vos, W. M. Unravelling lactate-acetate and sugar conversion into butyrate by intestinal anaerobutyricum and Anaerostipes species by comparative proteogenomics. Environ. Microbiol. 22, 4863–4875. https://doi.org/10.1111/1462-2920.15269 (2020).
Henke, M. T. et al. Ruminococcus gnavus, a member of the human gut Microbiome associated with crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. U S A. 116, 12672–12677. https://doi.org/10.1073/pnas.1904099116 (2019).
Bell, A. et al. Elucidation of a Sialic acid metabolism pathway in mucus-foraging Ruminococcus gnavus unravels mechanisms of bacterial adaptation to the gut. Nat. Microbiol. 4, 2393–2404. https://doi.org/10.1038/s41564-019-0590-7 (2019).
Shin, A., Xu, H. & Imperiale, T. F. Associations of chronic diarrhoea with non-alcoholic fatty liver disease and obesity-related disorders among US adults. BMJ Open. Gastroenterol. 6, e000322. https://doi.org/10.1136/bmjgast-2019-000322 (2019).
Hehemann, J. H. et al. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912. https://doi.org/10.1038/nature08937 (2010).
Hehemann, J. H., Kelly, A. G., Pudlo, N. A., Martens, E. C. & Boraston, A. B. Bacteria of the human gut Microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc. Natl. Acad. Sci. U S A. 109, 19786–19791. https://doi.org/10.1073/pnas.1211002109 (2012).
De Filippis, F. et al. Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe 25, 444–453 e443 (2019). https://doi.org/10.1016/j.chom.2019.01.004
Derrien, M., Turroni, F., Ventura, M. & van Sinderen, D. Insights into endogenous bifidobacterium species in the human gut microbiota during adulthood. Trends Microbiol. 30, 940–947. https://doi.org/10.1016/j.tim.2022.04.004 (2022).
Jacobs, J. P. et al. Multi-omics profiles of the intestinal Microbiome in irritable bowel syndrome and its bowel habit subtypes. Microbiome 11, 5. https://doi.org/10.1186/s40168-022-01450-5 (2023).
Klem, F. et al. Prevalence, risk factors, and outcomes of irritable bowel syndrome after infectious enteritis: A systematic review and Meta-analysis. Gastroenterology 152, 1042–1054. https://doi.org/10.1053/j.gastro.2016.12.039 (2017). e1041.
Breen-Lyles, M., Decuir, M., Byale, A., Smith, K. & Grover, M. Impact of Rome IV criteria on the prevalence of post-infection irritable bowel syndrome. Neurogastroenterol Motil. 35, e14532. https://doi.org/10.1111/nmo.14532 (2023).
Pimentel, M. et al. Antibiotic treatment of constipation-predominant irritable bowel syndrome. Dig. Dis. Sci. 59, 1278–1285. https://doi.org/10.1007/s10620-014-3157-8 (2014).
Pimentel, M. et al. Rifaximin therapy for patients with irritable bowel syndrome without constipation. N Engl. J. Med. 364, 22–32. https://doi.org/10.1056/NEJMoa1004409 (2011).
Lembo, A. et al. N. AGA clinical practice guideline on the Pharmacological management of irritable bowel syndrome with diarrhea. Gastroenterology 163, 137–151. https://doi.org/10.1053/j.gastro.2022.04.017 (2022).
Vacca, M. et al. The controversial role of human gut lachnospiraceae. Microorganisms 8 https://doi.org/10.3390/microorganisms8040573 (2020).
Wang, Z. et al. Xylan alleviates dietary fiber deprivation-induced dysbiosis by selectively promoting bifidobacterium pseudocatenulatum in pigs. Microbiome 9, 227. https://doi.org/10.1186/s40168-021-01175-x (2021).
Choo, C. et al. Colonic mucosal microbiota is associated with bowel habit subtype and abdominal pain in patients with irritable bowel syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 323, G134–G143. https://doi.org/10.1152/ajpgi.00352.2021 (2022).
Louis, P. & Flint, H. J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19, 29–41. https://doi.org/10.1111/1462-2920.13589 (2017).
Jiang, L. et al. A high-fiber diet synergizes with prevotella Copri and exacerbates rheumatoid arthritis. Cell. Mol. Immunol. 19, 1414–1424. https://doi.org/10.1038/s41423-022-00934-6 (2022).
Pichler, M. J. et al. Butyrate producing colonic clostridiales metabolise human milk oligosaccharides and cross feed on mucin via conserved pathways. Nat. Commun. 11, 3285. https://doi.org/10.1038/s41467-020-17075-x (2020).
Wijdeveld, M. et al. Intestinal acetate and butyrate availability is associated with glucose metabolism in healthy individuals. iScience 26, 108478. https://doi.org/10.1016/j.isci.2023.108478 (2023).
Carco, C. et al. Increasing evidence that irritable bowel syndrome and functional Gastrointestinal disorders have a microbial pathogenesis. Front. Cell. Infect. Microbiol. 10, 468. https://doi.org/10.3389/fcimb.2020.00468 (2020).
Mulder, D. et al. Gut microbiota composition links to variation in functional domains across psychiatric disorders. Brain Behav. Immun. 120, 275–287. https://doi.org/10.1016/j.bbi.2024.05.037 (2024).
Yu, L., Chen, X., Bai, X., Fang, J. & Sui, M. Microbiota alters and its correlation with molecular regulation underlying depression in PCOS patients. Mol. Neurobiol. https://doi.org/10.1007/s12035-023-03744-7 (2023).
Xiao, L. et al. The interactions between host genome and gut Microbiome increase the risk of psychiatric disorders: Mendelian randomization and biological annotation. Brain Behav. Immun. 113, 389–400. https://doi.org/10.1016/j.bbi.2023.08.003 (2023).
Calderon, G. et al. Associations of habitual dietary intake with fecal Short-Chain fatty acids and bowel functions in irritable bowel syndrome. J. Clin. Gastroenterol. 56, 234–242. https://doi.org/10.1097/MCG.0000000000001521 (2022).
Wright, R. J., Comeau, A. M. & Langille, M. G. I. From defaults to databases: parameter and database choice dramatically impact the performance of metagenomic taxonomic classification tools. Microb. Genom. 9 https://doi.org/10.1099/mgen.0.000949 (2023).
Francis, C. Y., Morris, J. & Whorwell, P. J. The irritable bowel severity scoring system: a simple method of monitoring irritable bowel syndrome and its progress. Aliment. Pharmacol. Ther. 11, 395–402. https://doi.org/10.1046/j.1365-2036.1997.142318000.x (1997).
Mearin, F. et al. Bowel Disorders Gastroenterology https://doi.org/10.1053/j.gastro.2016.02.031 (2016).
Mulligan, A. A. et al. A new tool for converting food frequency questionnaire data into nutrient and food group values: FETA research methods and availability. BMJ Open. 4, e004503. https://doi.org/10.1136/bmjopen-2013-004503 (2014).
Heaton, K. W. et al. Defecation frequency and timing, and stool form in the general population: a prospective study. Gut 33, 818–824. https://doi.org/10.1136/gut.33.6.818 (1992).
Sadik, R., Abrahamsson, H., Ung, K. A. & Stotzer, P. O. Accelerated regional bowel transit and overweight shown in idiopathic bile acid malabsorption. Am. J. Gastroenterol. 99, 711–718. https://doi.org/10.1111/j.1572-0241.2004.04139.x (2004).
Park, J. et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 8, 80–93. https://doi.org/10.1038/mi.2014.44 (2015).
Tagliacozzi, D. et al. Quantitative analysis of bile acids in human plasma by liquid chromatography-electrospray tandem mass spectrometry: a simple and rapid one-step method. Clin. Chem. Lab. Med. 41, 1633–1641. https://doi.org/10.1515/CCLM.2003.247 (2003).
Vijayvargiya, P., Camilleri, M., Shin, A. & Saenger, A. Methods for diagnosis of bile acid malabsorption in clinical practice. Clin. Gastroenterol. Hepatol. 11, 1232–1239. https://doi.org/10.1016/j.cgh.2013.04.029 (2013).
Louis, P., Hold, G. L. & Flint, H. J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 12, 661–672. https://doi.org/10.1038/nrmicro3344 (2014).
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 8(4), e61217. https://doi.org/10.1371/journal.pone.0061217. PMID: 23630581; PMCID: PMC3632530 (2013).
vegan: Community Ecology Package v. 2.6-4. (2022).
Kanehisa, M. & Goto, S. K. E. G. G. Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2024).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Vieira-Silva, S. et al. Species-function relationships shape ecological properties of the human gut Microbiome. Nat. Microbiol. 1, 16088. https://doi.org/10.1038/nmicrobiol.2016.88 (2016).
Vujkovic-Cvijin, I. et al. Host variables confound gut microbiota studies of human disease. Nature 587, 448–454. https://doi.org/10.1038/s41586-020-2881-9 (2020).
Saad, R. J. et al. Do stool form and frequency correlate with whole-gut and colonic transit? Results from a multicenter study in constipated individuals and healthy controls. Am. J. Gastroenterol. 105, 403–411. https://doi.org/10.1038/ajg.2009.612 (2010).
Jaruvongvanich, V., Patcharatrakul, T. & Gonlachanvit, S. Prediction of delayed colonic transit using Bristol stool form and stool frequency in Eastern constipated patients: A difference from the West. J. Neurogastroenterol Motil. 23, 561–568. https://doi.org/10.5056/jnm17022 (2017).
Acknowledgements
The authors would like to thank the Mayo Clinic Department of Laboratory Medicine and Pathology and Purdue University Metabolite Profiling Facility for their collaboration and assistance with quantification of stool bile acids and short chain fatty acids.
Funding
AS has received funding from NIDDK K23DK122015 and R03DK132446.
Author information
Authors and Affiliations
Contributions
Developing study concept: AS. Planning study design: AS, HX, XG. Participant recruitment: AS, MRW, RS, TJS, NR, MB JW, CL, AG, JK, RA. Data collection and study procedures: AS, MRW, CL, AG, JK, ET. Data Management: AS, YX, MRW, CL, JK, HX, XG. Bayesian network analysis: AD. Data Analysis and Interpretation: YX, AD, XG. Drafting the manuscript: AS, XG. Critically revising the manuscript: MRW, RS, TJS, NR, MB, JW, ET, HX, AD.
Corresponding authors
Ethics declarations
Competing interests
AS serves as a consultant for Ardelyx, has served on an Advisory Board for Gemelli Biotech, and is an Advisor for Medis Labs, Inc.
Ethics approval and consent to participate
The study was approved by the Indiana University Institutional Review Board.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shin, A.S., Xing, Y., Waseem, M.R. et al. Microbiota and short chain fatty acid relationships underlie clinical heterogeneity and identify key microbial targets in irritable bowel syndrome (IBS). Sci Rep 15, 35375 (2025). https://doi.org/10.1038/s41598-025-19363-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-19363-2
