
Abstract
The GLUL gene encodes glutamine synthetase (GS), which plays a crucial role in glutamine–glutamate homeostasis. Both loss-of-function and gain-of-function variants of GLUL are known to cause genetic disorders in humans. Biallelic loss-of-function variants cause congenital glutamine deficiency, leading to developmental and epileptic encephalopathy (DEE) in an autosomal recessive manner. In contrast, certain variants of GLUL that lead to the loss of the N-terminal degron exert a gain-of-function effect, causing an autosomal dominant DEE. Only six autosomal recessive cases and ten autosomal dominant cases have been reported to date, and knowledge about GLUL-related DEE remains limited. In this study, we identified three unrelated patients with DEE carrying heterozygous de novo GLUL variants. One patient carried a variant that had been reported previously in two patients (c.-13–2A > G), and the other two patients carried novel candidate variants (c.-13-1G > C and c.604T > C). An alternative splicing event causing loss of the N-terminal degron of GS was confirmed by RNA sequencing in a patient carrying c.-13-1G > C variant. A comparison of our patients with previously reported cases revealed common symptoms, including epilepsy and global developmental delay. However, our patients exhibited additional phenotypes, such as hypertonia, cerebral atrophy, and T2 hyperintensity in deep grey matter, which have not been described in patients with autosomal dominant GLUL-related DEE. The seizure patterns and responses to antiseizure medications varied among patients, reflecting their diverse phenotypic spectrum. Similarly, biochemical analyses of plasma and cerebrospinal fluid showed heterogeneous profiles. We present analyses of the GLUL-related DEE with detailed clinical descriptions and identified novel causal variants. Comparative analysis of genotypes and phenotypes revealed the diverse nature of the disease, expanding our knowledge about the genetic and clinical spectrum of GLUL-related DEE.
Similar content being viewed by others


Introduction
Glutamine synthetase (GS), an enzyme encoded by the GLUL gene, plays a crucial role in the physiology of the human central nervous system by converting glutamate and ammonia into glutamine1,2. Alterations in the functionality of the protein can disrupt glutamate–glutamine homeostasis, causing severe consequences in brain development and function. Individuals with loss-of-function variants in both GLUL alleles exhibit congenital glutamine deficiency in an autosomal recessive (AR) manner (OMIM: 610015)3. To date, six patients have been reported to have this disease, with symptoms including global developmental delay (GDD), seizure, and hypotonia3,4,5,6,7.
Recently, developmental and epileptic encephalopathy (DEE) 116 (OMIM: 620806), which is an autosomal dominant (AD) disease caused by gain-of-function GLUL variants, was identified8. Ten patients have been reported to date, and all carried heterozygous variants in the start codon or canonical splicing site at 5’ untranslated region (UTR) of exon 2, which leads to the loss of normal translation start site8,9. Translation is then initiated at the alternative in-frame downstream start codon, methionine 18, producing a smaller GS with a truncated N-terminal region. Normally, the lysine residues located in the N-terminal region of GS are acetylated at high glutamine concentrations, triggering ubiquitin-mediated protein degradation and serving as a negative feedback mechanism10. However, the loss of this critical domain disturbs the regulatory process while preserving the enzyme activity, causing pathological symptoms including epilepsy and GDD8,9.
As an AD disease associated with the GLUL gene is a newly characterized disorder with a unique pathophysiology, little is known about its genetic and clinical spectrum. In this paper, we present three additional patients with heterozygous de novo GLUL variants, two of which were novel candidate variants. Moreover, we provide detailed descriptions of clinical courses and a comparative analysis of genotypes and phenotypes, incorporating all patients reported previously to expand the clinical knowledge of GLUL-related DEE.
Methods
We identified three patients (Patients 1, 2, and 3) carrying pathogenic or likely pathogenic GLUL variants. All patients were evaluated for neurodevelopmental problems and underwent molecular genetic testing at the Seoul National University Children’s Hospital. All of them underwent trio whole-exome sequencing, including the probands and their parents. Additionally, whole-genome sequencing was performed as a singleton for Patient 1 and as a trio for Patients 2 and 3. To facilitate further diagnostic evaluation, Patient 2 underwent bulk RNA sequencing (RNA-seq) from the muscle tissue and Patient 3 underwent confirmatory Sanger sequencing for the novel GLUL variant.
All sequencing was performed using Illumina technology, and sequence reads were aligned to the human reference genome hg38. In the case of exome and genome sequencing, subsequent downstream processing was based on the Genome Analysis Toolkit best practice workflows11. RNA-seq data were also aligned to hg38 using STAR in 2-pass mode12. The splicing events identified via RNA-seq were visualized using ggsashimi13. Public genome databases, including gnomAD14, BRAVO15, and KOVA16, as well as in-silico pathogenicity prediction tools, including SpliceAI17, REVEL18, CADD19, AlphaMissense20, phyloP21, and PhastCons22, were used for variant analysis.
Non-silent variants in exonic or splicing regions were prioritized and subsequently filtered based on population allele frequency. De novo variants were selected to evaluate possible AD or X-linked dominant inheritance, while homozygous, hemizygous, or compound heterozygous variants were selected for potential AR or X-linked recessive inheritance. Finally, variants in genes with known disease associations were prioritized for further analysis (Supplementary Fig. S1). Candidate variants were classified according to the standards and guidelines of the American College of Medical Genetics and Genomics23.
The medical records of the patients were reviewed regarding the description of their clinical courses. Each patient’s medical record was documented by their attending paediatric neurologist at Seoul National University Children’s Hospital, based on clinical judgment from the patient’s medical history and physical examination. Phenotypes including initial symptoms, seizure type and frequency, developmental status, and neurological examination results were collected sequentially. Prescribed antiseizure medication and the response to these treatments were reviewed to assess drug efficacy.
Results from laboratory tests, functional assessments, and imaging studies were also collected. Plasma amino acid analysis, including glutamine and glutamate levels, was performed for Patients 1, 2, and 3, and Patient 3 additionally underwent cerebrospinal fluid (CSF) amino acid analysis. Plasma ammonia levels were measured for all patients during outpatient visits. Electroencephalography (EEG) and brain magnetic resonance imaging (MRI) were performed for initial seizure evaluation and follow-up assessments.
Results
Identification of GLUL variants
The detailed coverage statistics of each sequencing are provided in Supplementary Table S1, and the process of variant filtration is summarized and illustrated in Supplementary Fig. S1. As a result, we identified three de novo GLUL variants in unrelated Korean patients with DEE. Two patients carried heterozygous variants in the canonical splicing acceptor site at the 5’ UTR of exon 2, c.-13–2A > G in Patient 1 and c.-13-1G > C in Patient 2. SpliceAI predicted the loss of the original acceptor site with a delta score of 1.00 for both variants, and a new acceptor site was predicted to be located 26 bp downstream of the original site. Neither of the variants was found in population databases including gnomAD, BRAVO, and the Korean-specific genomic database KOVA (Table 1).
Patient 3 carried a de novo missense variant, c.604T > C (p.Trp202Arg), as a novel candidate variant. The variant and its de novo status were confirmed by Sanger sequencing (Supplementary Fig. S2). This variant and the corresponding amino acid change were absent in the normal population databases. The variant locus was highly conserved among vertebrate species, and various in-silico predictors classified it as pathogenic (Table 1). Moreover, a whole-genome analysis did not identify any additional pathogenic or likely pathogenic variants. This variant was classified as likely pathogenic according to the following evidence: PS2 (De novo), PM2 (Absent from controls), and PP3 (In-silico prediction pathogenic).
Comparative and experimental analysis of GLUL variants
Figure 1A summarizes all variants identified in patients with GLUL-related DEE3,4,5,6,7,8,9, and the genotypes of individual patients are reported in Table 2. Most AR variants were missense, and all were found homozygous because of consanguinity except for c.42G > C (p.Lys14Asn), which was found in trans with a nonsense variant c.316C > T (p.Arg106Ter). All previously reported AD variants were clustered in the canonical splicing site or the canonical start codon of exon 2, as described previously.

Analysis of the identified GLUL variants. (A) Loci of the reported GLUL variants. The orange dots indicate the variants from our patients, and the blue dots denote previously reported variants. Variants assumed to cause autosomal dominant (AD) diseases are shown above the gene panel, and variants associated with autosomal recessive (AR) disease are shown under the panel. PhyloP scores for each genomic position are shown below, and the scores for the missense variants (p.Lys14Asn, p.Arg41Cys, p.Trp202Arg, p.Arg324Ser/p.Arg324Cys, and p.Arg341Cys) are highlighted. AD variants were clustered in the splicing site or the start codon, although one variant from our patient was a missense variant. The AR variants were all missense, with the exception of one truncating variant. (B) Bulk RNA sequencing (RNA-seq) result of Patient 2 carrying a novel c.-13-1G > C variant. The top panel shows the RNA-seq result of Patient 2, and the next two panels show the RNA-seq result of two normal controls. The numbers indicate the count of each splicing event. The lowest panel shows the MANE transcript of GLUL. An alternative splicing event causing the loss of the canonical start site in exon 2 was observed in Patient 2, whereas the controls did not show this event.
The analysis of our three patients identified two novel candidate variants (c.-13-1G > C and c.604T > C) and one variant that had been reported previously in two additional patients (c.-13–2A > G). The c.-13-1G > C variant is located in the canonical splicing site, and a different nucleotide change in the same position (c.-13-1G > A) was reported in one patient. Another novel variant, c.604T > C (p.Trp202Arg), was heterozygous without any additional pathogenic or likely pathogenic variant in GLUL, unlike all other previously reported missense variants which exhibited a biallelic status. An analysis of phyloP scores indicated that the locus was well-preserved, exhibiting the highest score (8.775) among all reported missense variants (Fig. 1A).
The RNA-seq results of Patient 2 were analysed to verify that the c.-13-1G > C variant caused an alternative splicing event consistent with the start–loss mechanism of other AD variants. The result revealed novel RNA transcripts with a missing start codon, which were not found in the normal control group (Fig. 1B). The new splicing site was located 26 bp downstream of the original site, skipping the original start codon as predicted by SpliceAI. The novel splicing event was observed in 35% (626/1781) of the total exon 1–2 junctions.
Clinical presentations
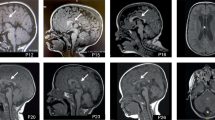
Patient 1 was a 6-year-old male referred to the medical genetics clinic for intractable epilepsy. He was born with a birth weight of 3.46 kg without any perinatal problems. He initially showed normal growth and development. At 3 months of age, he began to show seizure-like movements, and MRI revealed cerebral atrophy (Fig. 2A). The initial EEG results were consistent with infantile spasms. Treatment with vigabatrin and prednisolone was successful at that time, but the seizures re-emerged at 8 months of age.

Brain MRI findings of Patients 1, 2, and 3. (A–C) Serial brain MRI images of Patients 1, 2, and 3. The first and second rows show axial FLAIR images for (A and C) and axial T2 images for (B). The last row shows sagittal T1 images. (A) MRI image of Patient 1 taken at 3 months (left) and 5 years (right). The axial images show prominent perivascular spaces and high signal intensity in the thalamus. The sagittal images reveal dysmorphic corpus callosum. (B) MRI image of Patient 2 taken at 16 months (left) and 6 years (right). The axial images show prominent perivascular spaces with T2 hyperintensity in the bilateral basal ganglia and the thalamus. (C) MRI image of Patient 3 taken at 1 month (left) and 15 months (right). MRI performed at 15 months shows diffuse cerebral atrophy with a thin corpus callosum.
At 21 months of age, the patient exhibited generalized tonic–clonic seizures approximately once per week, despite treatment with topiramate, valproic acid, and lacosamide. Although the patient could control his head and babble, further developmental milestones were not achieved. Biochemical analysis revealed a borderline low plasma glutamine level, accompanied by an elevated ammonia level of 227 µg/dL. Sleep EEG showed diffuse delta activity in the background with a few centroparietal sharp waves. Seizure frequency decreased to once per month with the addition of perampanel at 4 years old. Follow-up MRI revealed symmetric T2 hyperintense lesions involving the thalamus, mammillary body, and brainstem with prominent perivascular spaces (Fig. 2A). At the last follow-up at 6 years of age, he was only able to sit with support, with no further developmental progress observed, suggesting developmental arrest.
Patient 2 was a 7-year-old male referred to the paediatric neurology clinic because of GDD. He was born at 38 weeks with a birth weight of 3.34 kg, without perinatal problems. His first seizure also occurred at 3 months of age, and by 16 months, he exhibited generalized tonic and myoclonic seizures approximately ten times per day. He was not able to control his head or speak any meaningful words. Brain MRI showed symmetric, ill-defined T2 high signal intensity in the bilateral basal ganglia, with prominent perivascular space at the white matter (Fig. 2B). Sleep EEG revealed diffuse high-amplitude polymorphic delta activity with posterior dominant spike-wave discharges. Plasma glutamine and ammonia levels were within the normal range.
Seizures persisted despite trials of multiple antiseizure medications and a ketogenic diet, but at 3 years of age, the seizures were controlled with the addition of valproic acid. The seizures recurred at 5 years of age after a febrile illness caused by pneumonia, with their frequency reaching up to 10 times per day at 6 years old. The seizures were initially stabilized after adding vigabatrin, but frequent relapses were observed despite the use of lamotrigine, levetiracetam, vigabatrin, and valproic acid. Although neurological examination revealed increased rigidity, the patient gained the ability to control his head and roll over. Nevertheless, no further developmental milestones were achieved even by 7 years of age. MRI showed cerebral atrophy with ventriculomegaly, along with T2 hyperintense lesions in the bilateral thalami and prominent perivascular spaces (Fig. 2B).
Patient 3 was a 4-year-old female who presented to the paediatric neurology clinic because of early-onset epilepsy. Generalized tonic seizures with facial flushing began at 4 weeks of age, which evolved into clonic seizures occurring approximately 10 times per day. MRI did not reveal any abnormalities in the brain (Fig. 2C). At 1 year of age, neurological examination revealed microcephaly and generalized spasticity with hyperreflexia. She was unable to control her head or make appropriate eye contact. Follow-up brain MRI revealed diffuse white matter volume loss with diffuse thinning of the corpus callosum (Fig. 2C), and interictal EEG showed diffuse rhythmic delta activity with frequent spike discharges in the left temporal region. The plasma and CSF glutamine levels were normal, although the ammonia level measured in an outpatient setting was elevated to 184 µg/dL.
She became seizure-free with valproic acid, topiramate, levetiracetam, clonazepam, and pyridoxine, but by age three, spasms-like seizures with sudden arousal and facial flushing re-emerged. By her last follow-up at 4 years of age, no further developmental milestones had been achieved. She continued to lack adequate head control, and microcephaly persisted with a head circumference of 43.5 cm (< 3rd percentile).
Phenotypic spectrum of GLUL-related DEE
Table 2 presents a summary of the clinical findings for the patients with GLUL-related DEE, incorporating data from 10 patients with AD type of the disease8,9 and 6 patients with AR type of the disease3,4,5,6,7 reported previously.
All patients shared common features including epilepsy and profound GDD. Most patients experienced refractory seizures, and a few treatments such as ketogenic diet, perampanel, valproic acid, vigabatrin, and brivaracetam exhibited partial efficacy in some patients. Although all patients carrying AD variants in the previous studies exhibited hypotonia, the neurological examinations of our patients revealed rigidity and spasticity. Uniquely, Patient 3 had microcephaly, which was only found in patients with AR type of the disease. Two patients from the AR group had died in the neonatal period, at 2 days for Patient 14 and 4 weeks for Patient 15.
The brain MRI findings consistently revealed prominent perivascular spaces and thinning of the corpus callosum in patients with AD type of the disease. Although hypomyelination or demyelination was also commonly observed in previously reported cases (7 out of 10 in AD cases), our patients did not have these features. Conversely, all our patients showed cerebral atrophy, and two of them (Patients 1 and 2) had T2 hyperintensity in the basal ganglia or the thalamus. Patient 3 did not have this feature, but the degree of cerebral atrophy and corpus callosum thinning were the most severe among our patients. In AR cases, abnormalities such as cerebral atrophy, attenuation of the gyri, and hypomyelination were found in Patients 14–16, whereas the MRI images of Patients 17–19 were structurally normal.
Although some patients with the AD type of the disease showed reduced plasma glutamine levels, most patients exhibited normal plasma glutamine. The CSF glutamine levels were also inconsistent among patients, ranging from low to high. Furthermore, Patients 1 and 3 in our cohort showed elevated ammonia levels. In AR cases, low plasma or CSF glutamine and elevated ammonia levels were common findings, although two patients (Patients 18 and 19) showed normal plasma glutamine levels.
Discussion
We identified three additional patients with DEE carrying GLUL variants, and two had novel causal variants. One was a splicing variant which was confirmed by RNA-seq to be consistent with the start–loss disease mechanism, as previously reported8, whereas the other one was a missense variant. We also described clinical presentations in detail, together with serial MRI findings, to provide additional information about disease progression and prognosis.
In the first study that introduced AD GLUL-related DEE, all patients were females, and the presence of a phenotypic difference between sexes caused by variations in glutamine metabolism was speculated8. However, along with one male patient reported recently9, our two additional male patients exhibited phenotypes which were comparable to that of the previously reported female patients. This finding suggests that sex is unlikely to affect disease pathogenesis significantly, thereby expanding our understanding of the disease manifestation.
All patients with GLUL-related disease exhibited both epilepsy and GDD. However, these are nonspecific features commonly observed in individuals with monogenic neurodevelopmental disorders, making it difficult to identify the causative gene based solely on clinical presentation. The phenotypic spectrum of this condition is highly variable, even among patients with the same mode of inheritance (AD and AR). Only a few antiseizure medications appeared to provide partial benefit, and treatment response varied markedly among patients. Accurate assessment of their efficacy is further complicated by the concurrent use of multiple agents.
Prominent perivascular spaces, which is a finding associated with small vessel disease and various neuroinflammatory and neurodegenerative conditions, were identified in most patients with AD type of the disease24. Cerebral atrophy and T2 hyperintensity in the bilateral deep grey matter were also commonly observed in our cohort. These findings are frequently associated with systemic metabolic abnormality and mitochondrial diseases25, and might reflect the crucial role of glutamine and glutamate in cell energy metabolism26,27. Further investigation is needed to understand the spectrum of the neuroimaging results of GLUL-related disease.
Since GS converts ammonia and glutamate to glutamine, loss-of-function of this enzyme is expected to result in decreased glutamine levels, accompanied by elevated ammonia and glutamate levels. Conversely, gain-of-function would be expected to cause the opposite pattern. Biochemical analyses in patients with AR type of the disease were generally consistent with the loss-of-function mechanism of GS. In contrast, the analysis of patients with AD type of the disease did not yield consistent results with the gain-of-function mechanism. Some showed low glutamine levels in the plasma or CSF and elevation of ammonia, although the high ammonia level might have been caused by the use of valproic acid28. These observations suggest that biochemical testing might have some value in confirming the AR type of the disease, but its value in the diagnosis of the AD type of the disease remains unclear.
In the AD disease group, only Patient 3 carried a missense variant (p.Trp202Arg). We screened for the possibility of missing variants using trio whole-exome and whole-genome sequencing, but no additional pathogenic or likely pathogenic variant was found. The high missense constraint score (Z = 3.23, o/e = 0.6 [0.54–0.66])14 of GLUL with the prediction of various in-silico tools supports the pathogenicity of this missense variant, and an additional explanation for the pathophysiology of the AD type of the disease beyond the start-loss mechanism is required. One explanation is that the missense variant might cause a gain-of-function effect like the start-loss variants, as occasionally observed in disease-causing mutations in other proteins29,30,31. Another possibility is the presence of a dominant-negative effect, which is a phenomenon common in homomeric protein complexes32. Because GS also forms a homomeric decamer in humans33, the missense variant might disrupt normal GS function. This could explain the unique phenotype of Patient 3 which resembled the AR type of the disease, such as microcephaly and severe structural abnormality in the brain.
The GLUL gene is also highly constrained against loss-of-function with LOEUF of 0.38 and pLI of 114, indicating haploinsufficiency. However, most patients with confirmed AR disease carried homozygous missense variants. These findings might indicate that most truncating variants in GLUL are lethal in the early developmental stage, and thus not found in normal populations or in the patients with GLUL-related disease. Missense variants can express residual enzyme activity3,8, leading to viability of the carriers. Differences in the residual activity could also explain the milder phenotype observed in some patients34. One exception was the two siblings (Patient 18, 19) with one truncating variant (p.Arg106Ter) and one missense variant (p.Lys14Asn), and the cause of the mild phenotype of the patients is unclear. Together with our patient carrying a missense variant, these observations suggest the existence of an unveiled mechanism underlying the pathophysiology of GLUL-related DEE. Additional experiments aimed at analysing the functional properties of these variants are needed to fully elucidate the mechanism of the disease.
Conclusion
We characterized the clinical phenotypes of GLUL-related DEE in detail and identified novel causal variants. A comparative analysis of the genetic and clinical spectrum revealed unknown aspects of the disease, highlighting its diverse nature. These findings would help make predictions about the clinical presentation and prognosis of patients with the GLUL-related DEE. Considering that the total number of patients remains limited, there might be much left to be found about the disease. Further identification of individuals with pathogenic GLUL variants, along with the analysis of genotypes and their functional consequences, is needed to establish a full understanding of GLUL-related disease.
Data availability
The dataset generated and/or analyzed during the current study was deposited in the NCBI GEO repository (http://www.ncbi.nlm.nih.gov/geo/) (GSE297730).
References
Bak, L. K., Schousboe, A. & Waagepetersen, H. S. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 98 (3), 641–653 (2006).
Andersen, J. V. et al. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 196, 108719 (2021).
Haberle, J. et al. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl. J. Med. 353 (18), 1926–1933 (2005).
Haberle, J., Shahbeck, N., Ibrahim, K., Hoffmann, G. F. & Ben-Omran, T. Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol. Genet. Metab. 103 (1), 89–91 (2011).
Haberle, J. et al. Glutamine supplementation in a child with inherited GS deficiency improves the clinical status and partially corrects the peripheral and central amino acid imbalance. Orphanet J. Rare Dis. 7, 48 (2012).
Unal, O., Ceylaner, S. & Akin, R. A very rare etiology of hypotonia and seizures: congenital glutamine synthetase deficiency. Neuropediatrics 50 (1), 51–53 (2019).
Bennett, J., Gilkes, C., Klassen, K., Kerr, M. & Khan, A. Two siblings with Valproate-Related hyperammonemia and novel mutations in glutamine synthetase (GLUL) treated with carglumic acid. Child. Neurol. Open. 7, 2329048X20967880 (2020).
Jones, A. G. et al. Clustered de Novo start-loss variants in GLUL result in a developmental and epileptic encephalopathy via stabilization of glutamine synthetase. Am. J. Hum. Genet. 111 (4), 729–741 (2024).
Carbonell, E. et al. Male proband with intractable seizures and a de Novo start codon disrupting variant in GLUL. HGG Adv. 6(2), 100419 (2025).
Nguyen, T. V. et al. Glutamine triggers Acetylation-Dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol. Cell. 61 (6), 809–820 (2016).
McKenna, A. et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303 (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21 (2013).
Garrido-Martin, D., Palumbo, E., Guigo, R. & Breschi, A. Ggsashimi: Sashimi plot revised for browser- and annotation-independent splicing visualization. PLoS Comput. Biol. 14 (8), e1006360 (2018).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625 (7993), 92–100 (2024).
NHLBI UoMa. The NHLBI Trans-Omics for Precision Medicine (TOPMed) Whole Genome Sequencing Program. BRAVO variant browser 2018 [Available from: https://bravo.sph.umich.edu/freeze5/hg38/
Lee, J. et al. A database of 5305 healthy Korean individuals reveals genetic and clinical implications for an East Asian population. Exp. Mol. Med. 54 (11), 1862–1871 (2022).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176 (3), 535–548 (2019). e24.
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99 (4), 877–885 (2016).
Schubach, M., Maass, T., Nazaretyan, L., Roner, S. & Kircher, M. CADD v1.7: using protein Language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 52 (D1), D1143–D54 (2024).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with alphamissense. Science 381 (6664), eadg7492 (2023).
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R. & Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20 (1), 110–121 (2010).
Siepel, A. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15 (8), 1034–1050 (2005).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424 (2015).
Wardlaw, J. M. et al. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat. Rev. Neurol. 16 (3), 137–153 (2020).
Hegde, A. N., Mohan, S., Lath, N. & Lim, C. C. Differential diagnosis for bilateral abnormalities of the basal ganglia and thalamus. Radiographics 31 (1), 5–30 (2011).
Yoo, H. C., Yu, Y. C., Sung, Y. & Han, J. M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 52 (9), 1496–1516 (2020).
Bornstein, R., Mulholland, M. T., Sedensky, M., Morgan, P. & Johnson, S. C. Glutamine metabolism in diseases associated with mitochondrial dysfunction. Mol. Cell. Neurosci. 126, 103887 (2023).
Wong, Y. J., Mihic, T., Wan, A., Fan, J. & Gnyra, M. Valproic Acid-Associated hyperammonemia: A systematic Review-Reply to comments by Dr Aney. J. Clin. Psychopharmacol. 44 (1), 73–74 (2024).
Zweier, C., Sticht, H., Aydin-Yaylagul, I., Campbell, C. E. & Rauch, A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am. J. Hum. Genet. 80 (3), 510–517 (2007).
Rigter, P. M. F. et al. Role of CAMK2D in neurodevelopment and associated conditions. Am. J. Hum. Genet. 111 (2), 364–382 (2024).
Nil, Z. et al. Rare de Novo gain-of-function missense variants in DOT1L are associated with developmental delay and congenital anomalies. Am. J. Hum. Genet. 110 (11), 1919–1937 (2023).
Bergendahl, L. T. et al. The role of protein complexes in human genetic disease. Protein Sci. 28 (8), 1400–1411 (2019).
Tecson, M. C. B., Geluz, C., Cruz, Y. & Greene, E. R. Glutamine synthetase: diverse regulation and functions of an ancient enzyme. Biochemistry 64 (3), 547–554 (2025).
Frieg, B. et al. Molecular mechanisms of glutamine synthetase mutations that lead to clinically relevant pathologies. PLoS Comput. Biol. 12 (2), e1004693 (2016).
Acknowledgements
Not applicable.
Funding
This research was supported and funded by SNUH Lee Kun-hee Child Cancer & Rare Disease Project, Republic of Korea (grant number: 25B-001-0100).
Author information
Authors and Affiliations
Contributions
Conceptualization: D.E.O., S.L., and J.-H.C.; methodology: D.E.O., and S.L.; investigation: D.E.O.; validation: S.L.; formal analysis: D.E.O., S.S.J., and S.L.; data curation: D.E.O., S.S.J., and S.L.; writing – original draft preparation: D.E.O., S.L.; writing – review and editing: S.S.J., W.J.K., S.Y.K., B.C.L., K.J.K., and J.-H.C.; visualization: D.E.O., S.L.; supervision: W.J.K., S.Y.K., B.C.L., K.J.K.; funding acquisition: J.-H.C. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of Seoul National University Hospital (IRB No. 2412-122-1599). Informed consent was obtained from a parent and/or legal guardian for all study participants prior to blood sampling, and all methods were performed in accordance with relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oh, D.E., Jang, S.S., Kim, W.J. et al. Expanding the clinical and genetic spectrum of GLUL-related developmental and epileptic encephalopathy. Sci Rep 15, 35655 (2025). https://doi.org/10.1038/s41598-025-19666-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-19666-4
